
Abstract
Central nervous system cholinergic neurons arise from several discrete sources, project to multiple brain regions, and exert specific effects on reward, learning, and memory. These processes are critical for the development and persistence of addictive disorders. Although other neurotransmitters, including dopamine, glutamate, and serotonin, have been the primary focus of drug research to date, a growing preclinical literature reveals a critical role of acetylcholine (ACh) in the experience and progression of drug use. This review will present and integrate the findings regarding the role of ACh in drug dependence, with a primary focus on cocaine and the muscarinic ACh system. Mesostriatal ACh appears to mediate reinforcement through its effect on reward, satiation, and aversion, and chronic cocaine administration produces neuroadaptive changes in the striatum. ACh is further involved in the acquisition of conditional associations that underlie cocaine self-administration and context-dependent sensitization, the acquisition of associations in conditioned learning, and drug procurement through its effects on arousal and attention. Long-term cocaine use may induce neuronal alterations in the brain that affect the ACh system and impair executive function, possibly contributing to the disruptions in decision making that characterize this population. These primarily preclinical studies suggest that ACh exerts a myriad of effects on the addictive process and that persistent changes to the ACh system following chronic drug use may exacerbate the risk of relapse during recovery. Ultimately, ACh modulation may be a potential target for pharmacological treatment interventions in cocaine-addicted subjects. However, the complicated neurocircuitry of the cholinergic system, the multiple ACh receptor subtypes, the confluence of excitatory and inhibitory ACh inputs, and the unique properties of the striatal cholinergic interneurons suggest that a precise target of cholinergic manipulation will be required to impact substance use in the clinical population.
Similar content being viewed by others
INTRODUCTION
Insights from animal models and neuroimaging studies have led to a greater understanding of the addictive process. Dopamine (DA) has been identified as the critical neurotransmitter in the reward circuitry mediating substance abuse and the primary focus of preclinical research and clinical treatment interventions (Adinoff, 2004; Di Chiara et al, 2004; Self, 2004). DA levels abruptly increase following the administration of many drugs of abuse, including cocaine, and cocaine is no longer self-administered in animal models of addiction following DA receptor blockade within the nucleus accumbens (NAc) (Chang et al, 1994). However, the addictive effects of cocaine are not entirely attributable to DA. For example, interventions augmenting DA activity have generally not been successful in significantly ameliorating cocaine use in cocaine-dependent populations (Kleber, 2003; Kosten et al, 2002). DA's contribution to the neurobiological etiology of cocaine addiction, therefore, is most likely shared with multiple other neurotransmitter systems (Arnold, 2005; Bardo, 1998; Gorelick et al, 2004; Kalivas, 2004; Muller et al, 2003; Roberts, 2005).
Drug addiction has been described as a disease of the brain reward system wherein drugs activate the neuronal circuitry involved in reward and memory (Dackis and O’Brien, 2001). This activation produces an aberrant engagement of the learning process (Hyman, 2005). Because of the effect of cholinergic systems on reward and drug self-administration, the prevalence of acetylcholine (ACh) within the striatum (Butcher and Butcher, 1974; Pisani et al, 2001), and the involvement of ACh in higher order cognitive processes (Pepeu and Giovannini, 2004), ACh may play an important role in the addictive processes underlying cocaine dependence. To date, research has emphasized the role of ACh in neurocognitive disorders, such as Alzheimer's and Parkinson's disease (Bosboom et al, 2003; Mesulam, 1996), as well as nicotine dependence (Stolerman and Shoaib, 1991), with less attention devoted to the role of ACh in other addictive disorders.
This review will present and integrate the research on the role of ACh in cocaine dependence. We will initially provide an overview of the animal models of addiction described in this review, followed by an overview of the ACh system, highlighting aspects that are central to this paper. A discussion of the effects of reward, including cocaine, on mesostriatal ACh will follow. This will be coupled with a description of the effects of cholinergic perturbations on cocaine self-administration, reinforcement, and sensitization, as well as the effects of chronic cocaine administration on ACh receptors. The balance of the review will examine the role of hippocampal, striatal, amygdalar, and prefrontal ACh in various cognitive aspects of addiction, including learning, attention, and executive functioning, as these processes are believed to be crucial to the long-term maintenance of cocaine dependence. The brain regions associated with each aspect of the review are described and, where applicable, relevant neurobiologic disruptions in addicted subjects are discussed. The review concludes with a brief discussion of the potential of ACh as a target of pharmacological treatment intervention and future directions of study. A table (Table 1) provides a summary of the key conclusions and references described in this review.
OVERVIEW OF ANIMAL MODELS OF ADDICTION
Various animal model paradigms have been utilized in elucidating the role of ACh in addiction, most notably self-stimulation, self-administration, drug reinstatement, and conditioned place preference (CPP) (O’Brien and Gardner, 2005; Sanchis-Segura and Spanagel, 2006). In self-stimulation studies, experimental animals perform an operant response to electrically stimulate a specific brain region (Olds and Milner, 1954). Positioning the electrode within the ventral tegmental area (VTA) or DA projections (mesostriatal pathway) to the NAc produces the most reliable intracranial self-stimulation (ICSS) response rate (Wise, 1996). The most commonly used ICSS method is the curve shift paradigm (Miliaressis et al, 1986). By altering the ICSS current, the resulting response rate curve resembles the traditional dose–response curves obtained in pharmacology. The drug effects on reinforcement can then be estimated by comparing ICSS curves from saline and drug sessions. Within the mesostriatal pathway, DA agonists increase the effects of the ICSS reinforcers and produce the same response rate with lower ICSS current values (leftward shift), while DA antagonists have the opposite effect (Miliaressis et al, 1986).
In self-administration studies, animals are trained to perform a response (eg lever press or nose poke) that results in administration of the drug being studied. The reinforcing qualities of the drug are gauged by varying the number of responses required to obtain a drug dose. The more responses an animal provides to obtain a drug, the more reinforcing the drug (Gardner, 2000; Jacobs et al, 2003). Fixed ratio (FR) models of reinforcement are often used to assess drug effects. In FR models, animals increase their rate of self-administration as the unit dose of a drug is decreased (to compensate for decreases in the unit dose) and reduce responding as the unit dose is increased (Arnold and Roberts, 1997; Koob, 2000). Progressive ratio schedules are used to evaluate the reinforcing efficacy of a self-administered drug by increasing the response requirements for each successive reinforcement and determining the breaking point at which the animal will no longer respond (Arnold and Roberts, 1997; Koob, 2000). Increasing the unit dose of drug self-administration increases the breaking point on a progressive schedule. Another factor to consider in interpreting results is the effect of the inverted U-shaped dose–response curve on responding (Koob, 2000). A decrease in the number of reinforcements obtained may reflect either a decrease or an increase in the drug's reinforcing effects, depending on the doses of drug chosen for study.
In reinstatement models, the extinction of drug self-administration is followed by re-exposure to experimenter-administered drugs, drug-associated stimuli, or stressors. These procedures reinstate the drug-seeking behaviors that previously resulted in drug administration (eg lever press). This paradigm has been widely applied to assess the psychological and neurobiological mechanisms of relapse (Katz and Higgins, 2003; Schindler et al, 2002; Shaham et al, 2003). In the CPP paradigm, the rewarding properties of a compound are associated with the particular characteristics of a given environment (Bardo et al, 1995; Schechter and Calcagnetti, 1993). After conditioning, the animal spends more time in the environment associated with the rewarding drug. When the CPP paradigm is used in the context of sensitization, animals experience a greater drug effect in a previously conditioned, vs a novel, location (Robinson et al, 1998; Stewart and Vezina, 1988).
Useful strains of mice (ie ‘knockout mice’) have also been developed by genetically disrupting drug targets (receptors and transporters) or proteins in the target's pathway (Balster, 1991). Although most of the referenced studies are in vivo by design, in vitro studies have also been used to isolate various biological mechanisms from the living organism (Bolanos et al, 2000, 2002; Vanderschuren et al, 1999).
OVERVIEW OF THE CENTRAL CHOLINERGIC SYSTEM
Central nervous system (CNS) cholinergic neurons arise from multiple discrete sources, projecting to specific brain regions with well-defined cognitive, affective, and behavioral functions. Within the CNS, ACh is involved in motor behaviors, modulating the behavioral states associated with incoming information such as emotional tone, motivation, and arousal (Mesulam, 1996), as well as complex cognitive processes such as attention, learning, and memory (Sarter et al, 2003). Limbic and paralimbic regions of the CNS contain the highest density of ACh innervations in the brain (Mesulam, 1996) and are thought to be most relevant to the process of addiction (Bonson et al, 2002; Childress et al, 1999; Goldstein and Volkow, 2002). These regions include the ventral striatum (including the NAc), dorsal striatum, VTA, substantia nigra (SN), amygdala, hippocampus, and prefrontal cortex (PFC) (Lautin, 2001; Papez, 1995).
Muscarinic and Nicotinic ACh Receptors
The effects of ACh are mediated by muscarinic (mAChR) and nicotinic (nAChR) receptors. mAChRs are slow-acting, G-protein-coupled receptors that mediate their responses by activating a cascade of intracellular pathways. Activation of mAChRs by an applicable agonist causes a prolonged reduction of potassium conductance, inducing heightened cortical receptivity to other excitatory input (Wess, 1993). In contrast, nAChRs are fast-acting, ligand-gated ion channels that, upon binding with ACh, open to allow the diffusion of cations (Mihailescu and Drucker-Colin, 2000).
Molecular cloning studies have revealed the existence of five distinct mAChR subtypes (M1–M5). These receptors are widely expressed throughout the central and peripheral nervous system (Wess, 1993). M1, M2, and M4 AChRs are mainly present in forebrain regions, while M3 and M5 receptors are distributed throughout the brain (Bonner et al, 1987; Levey, 1996). M1, M3, and M5 (M1-like) receptors are coupled to Gq proteins that activate phospholipases. In contrast, M2 and M4 receptors (M2-like) are coupled to Gi proteins and inhibit adenylyl cyclase. Within the mesostriatal pathway, M5 receptors are primarily found in the midbrain (Weiner et al, 1990), whereas M1 and M4 (and some M2) are in the striatum (Levey et al, 1991). Neuronal nicotinic receptor subtypes include α and β subunits loosely categorized as two types: α–β subunit combinations (α2–α6, α10, and β2–β4) and homo-oligomeric combinations (α7–α9). Of the former group, α2β4 are the most common subunits and are widely expressed in the mammalian brain, including the dopaminergic midbrain and striatal regions. Of the latter group, only the α7 subunit is widely distributed in the mammalian brain (Graham et al, 2002; Zhou et al, 2003).
ACh Interneurons and Limbic Pathways
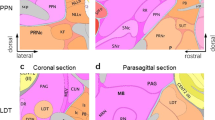
ACh interneurons are the primary source of ACh to the dorsal and ventral striatum (Calabresi et al, 2000; Pisani et al, 2001; Zhou et al, 2002). These interneurons integrate a variety of cognitive, limbic, and motor information (Tisch et al, 2004) and undergo plasticity and learning, which, in turn, influences striatal output signaling (Aosaki et al, 1994). In addition to striatal interneurons, eight discrete ACh neuronal pathways innervate the functional subdivisions of the human cerebral cortex and subcortical structures of the CNS (Mesulam, 1996). Three of these pathways are particularly relevant to this discussion (see Figure 1): (1) the mesopontine nuclei (comprised of the pedunculopontine and laterodorsal tegmental nuclei) of the rostral brainstem, which provides ACh innervations to the VTA, SN, and thalamus (Mena-Segovia et al, 2005); (2) the nucleus basalis of Meynert (NBM), which provides the principal ACh input to the cerebral cortex and amygdala (Mesulam, 1996); and (3) the medial septal-diagonal band of Broca, which provides primary ACh input to the hippocampus (Lewis and Shute, 1967). For purposes of this review, the NBM and medial septal-diagonal band are at times collectively referred to as the basal forebrain.

Limbic and paralimbic regions contain the highest density of acetylcholine (ACh) innervations in the brain. The three primary sources of cholinergic input are (1) the mesopontine nuclei (comprised of the pedunculopontine and laterodorsal tegmental nuclei), which provide ACh innervations to the VTA, SN, and thalamus, (2) the nucleus basalis of Meynert (NBM), which provides the principal ACh input to the cerebral cortex and amygdala, and (3) the medial septal-diagonal band of Broca, which provides primary ACh input to the hippocampus. Striatal ACh interneurons are largely influenced by dopamine (DA) receptors D1 and D2. Striatal muscarinic ACh interneurons primarily consist of M1, M2, and M4; M1 is post-synaptic (Mpost in the figure) and excitatory whereas M2 and M4 are pre-synaptic and inhibitory (Mpre in the figure). These interneurons synapse with γ-aminobutyric acid (GABA) medium spiny output neurons (MSNs). The ventral striatum, central to the motivations and reward behaviors that underlie drug addiction, projects output neurons to the ventral pallidium (VP) of the globus pallidus (GP) and, in turn, to the mediodorsal (MD) nucleus of the thalamus. The dorsal striatum, involved in the motor processes and conditioned learning of drug addiction, sends projections either directly or indirectly (via the external globus pallidus and subthalamic nucleus) to the internal segment of the globus pallidus and pars recticula of the substania nigra. The GP further projects, through GABAergic neurons, to the mediodorsal (MD) nucleus of the thalamus. Glutaminergic neurons from the MD project to the prefrontal cortex (PFC). Synaptic communication within the reward circuit is conducted via DA (modulatory), acetylcholine (ACh, modulatory), glutamate (GLU, excitatory), and γ-aminobutyric acid (GABA, inhibitory) neurotransmission.
MESOSTRIATAL ACETYLCHOLINE AND REWARD MEDIATION
The reward system is engaged in basic survival functions such as hunger, thirst, and sexual arousal (Hyman, 2005; Shizgal et al, 2001). These motivational states increase the salience of the goal objects (food, water, sexual partner), increasing the likelihood that these rewards are actively sought (Kelley and Berridge, 2002). Like natural rewards, addictive drugs, such as cocaine, stimulate the reward pathway and produce subjective feelings of pleasure. However, when drugs of abuse are repeatedly used, behaviors related to sustaining the addiction may progressively supplant behaviors optimal for survival. This single-minded pursuit tends to undermine, rather than promote, species survival, effectively ‘hijacking’ the neural systems related to the pursuit of rewards (Hyman et al, 2006).
The rewarding effects of natural reinforcers and addictive drugs, including cocaine, are related to their ability to activate the mesolimbic circuit in the midbrain VTA and stimulate the release of DA within the shell compartment of the NAc (Caine et al, 1997; Wise and Rompre, 1989). Drugs that facilitate DA transmission within the mesolimbic pathway enhance the processes by which otherwise neutral stimuli acquire incentive salience and motivate further drug-seeking behavior (Berridge and Robinson, 1998; Wyvell and Berridge, 2000). Both natural rewards and addictive drugs initially stimulate the release of DA from the mesolimbic pathway. Cocaine elevates DA levels in the NAc by blocking DA transporter-mediated reuptake, prolonging the time that DA remains in the synaptic cleft. This results in increased synaptic concentrations of DA, which binds with post-synaptic DA receptors (Kupfermann et al, 2000). The DA response to a natural reward does not generally persist over time, as the organism rapidly habituates to these stimuli after a few exposures. Conversely, the response to addictive drugs is not influenced by habituation, as each drug administration stimulates the release of DA (Bradberry et al, 2000; Di Chiara et al, 2004; Lecca et al, 2007). In addition, the DA response of addictive substances is several fold higher than that elicited by novel, naturally occurring rewards (Di Chiara et al, 1999).
Mesostriatal Cholinergic Neurons
ACh projections from the laterodorsal tegmental nucleus of the pons activate VTA DA neurons via ACh receptors. Both muscarinic, particularly M5 (Weiner et al, 1990), and nicotinic (Clarke and Pert, 1985) receptors provide input into VTA dopaminergic neurons (DANs). In turn, afferent DA projections from the VTA enervate the shell compartment of the NAc and activate ACh striatal interneurons, initiating the release of ACh. In the striatum, M1, M2, and M4 receptor subtypes predominate, whereas M3 and M5 subtypes are barely detectible (Weiner et al, 1990). The striatal cholinergic interneurons are physically large but make up less than 5% of the striatal neurons (the others being GABAergic medium spiny neurons (MSNs) and GABAergic interneurons) and function through both direct synaptic transmission and volume transmission (spillover) (Koos and Tepper, 2002). Striatal ACh interneurons are tonically active neurons (TANs); these neurons exhibit a transient depression of tonic firing in response to both rewarding and aversive environment events and are influenced by past experience (Apicella, 2007). Specifically, these interneurons appear highly responsive to stimulus detection, movement control, and context recognition. Thus, while TANs are somewhat responsive to the experience of rewards, they exhibit a significantly greater response to contextual features (cues) associated with the reward (Aosaki et al, 1994; Apicella et al, 1997; Kimura et al, 2003). This feature distinguishes the TANs from mesostriatal DANs, which are equally responsive to both rewards and their associated cues. In addition, TANs do not show a differential response based on reward probability whereas striatal DA activation reflects a mismatch between expectation and outcome (Morris et al, 2004). (These findings are best described for cholinergic interneurons in the dorsal striatum.) TANs synapse with striatal GABAergic MSNs. In contrast to the striatal ACh interneurons, the MSNs are physically active neurons (Apicella, 2002). The GABAergic MSNs project back to ACh interneurons and to basal forebrain ACh neurons (in addition to other regions), which, in turn, project to the cerebral cortex.
ACh and DA systems appear to coordinate striatal reward function in a feed-forward, complementary manner (Zhou et al, 2003). Striatal ACh is primarily controlled by VTA D1-like (D1, D5) and D2-like (D2, D3, D4) receptors. Both D1- and D2-like receptors have been localized on the somata, dendrites, and axons of striatal cholinergic neurons (Alcantara et al, 2003; Berlanga et al, 2005). Activation of the D1 receptor subtypes stimulates, whereas that of D2 receptor subtypes inhibits, striatal ACh release (Berlanga et al, 2005; Bertorelli and Consolo, 1990; Consolo et al, 1999; Drukarch et al, 1990; Stoof et al, 1987). Zhang et al (2002) have suggested that the offsetting action of the D1 and D2 receptor subtypes maintains the balance between ACh and DA. For example, whereas the dopaminergic stimulants cocaine and d-amphetamine and the D1 agonist SKF 82958 increased ACh in the NAc shell and core, the selective D1 SCH 39166 antagonist decreased accumbens ACh. The NAc ACh release induced by d-amphetamine, cocaine, and SKF 82958 was antagonized by SCH 39166, suggesting that D1 exerts tonic stimulatory control (Consolo et al, 1999). However, in an elegant study in baboons utilizing positron emission tomography (PET), Ding et al (2000) assessed striatal nicotinic ACh output following the administration of D1 and D2 agonists (D1: SKF 38393; D2: quinpirole) and antagonists (D1: SCH 23390; D2: raclopride). Nicotinic receptor binding was determined using the selective ACh receptor ligand norchloro[18F]fluoroepibatidine ([18F]NFEP). Striatal [18F]NFEP binding was increased following pretreatment with the D2 agonist and decreased following pretreatment with the D2 antagonist. Neither the D1 agonist nor antagonist altered striatal [18F]NFEP binding. Changes in striatal nicotinic AChR availability following D2, but not D1, agonist/antagonist challenges thus suggested that the D2 receptors predominantly influence striatal nicotinic ACh output under physiological conditions. Using almost identical DA agonists (D1: SKF 38393; D2: quinpirole) and antagonists (D1: SCH 23390; D2: sulpiride)) as Ding et al (2000), Deng et al (2007) measured dopaminergic inhibition of hyperpolarization-activated cation current in striatal cholinergic interneurons. As observed in the non-human primate study (Ding et al, 2000), D2 receptor, but not D1 receptor, compounds modulated the firing of these TANs (Deng et al, 2007).
Striatal cholinergic output, in turn, modulates striatal DA efflux (see Zhang et al, 2002). M4 receptors are often coexpressed with D1 receptors (Ince et al, 1997; Weiner et al, 1990), although these two receptors may initiate opposing actions. Zhang et al (2002) explored the relative influence of various mAChRs on DA output by assessing the effects of oxotremorine (an mAChR agonist) in rats selectively bred as M1, M2, M3, M4, and M5 deficients. The heightened efflux of DA induced by oxotremorine was unchanged in M1- and M2-deficient mice. M4 receptor-deficient mice demonstrated an absence of striatal DA output following oxotremorine, suggesting that M4 exerts a stimulatory effect on DA release. In contrast, M3-deficient mice showed an increase in DA release following oxotremorine, indicating that M3 receptors exert an inhibitory effect on DA. DA and ACh modulation is further enhanced by the distinct differences noted earlier in the specific stimuli that provoke striatal DA (DANs) and ACh (TANs) signaling (see Cragg, 2006).
Additional influences of striatal ACh include mAChRs located on striatal MSN output neurons. The MSNs induce the release of inhibitory neurotransmitter γ-aminobutyric acid, further promoting striatal signaling (see Figure 1). Harsing and Zigmond (1998) found that striatal GABAergic outflow is under the influence of M1 stimulatory and M3 inhibitory, but not M2, receptors, and Santiago and Potter (2001) have reported that M4 receptors are also highly expressed on GABAergic projection neurons. ACh also directly influences glutaminergic and GABAergic effects on midbrain DA neurons, further effecting striatal DA release (Grillner et al, 2000). High concentrations of mGlu2 receptors on striatal cholinergic neurons (Pisani et al, 2002), mediated through M2 and M3 receptors, reduce striatal glutaminergic release (Sugita et al, 1991). Serotonergic fibers from the raphe nucleus also demonstrate a potent excitatory effect on striatal cholinergic interneurons (Bonsi et al, 2007). Thus, as will become increasingly apparent in the following section, a myriad of inhibitory and excitatory DA and ACh receptor subtypes, coupled with complicated feedback interactions, result in relatively complex and at times inconsistent effects of either endogenous or exogenously administered ACh.
Non-dopaminergic Effects of Cocaine on ACh
Throughout this review, it will generally be presumed that the effects of cocaine on the cholinergic system are primarily mediated through dopaminergic efflux. Other direct interactions, however, may be in play. Cocaine has direct effects on both M1 and M2 (Flynn et al, 1992; Karpen and Hess, 1986; Sharkey et al, 1988), as well as nicotinic (Niu et al, 1995; Swanson and Albuquerque, 1987), cholinergic receptors. Cocaine also acts as a powerful local anesthetic through a direct effect on voltage-gated sodium channels. Recent work by Cooper et al (2006) suggests that the effect of cocaine on sodium channels in the subiculum may alter cocaine reinstatement. Both preclinical (DC Cooper et al, unpublished observation) and clinical (Adinoff et al, 2001) studies further suggest that neural activation by other sodium channel-mediated local anesthetics (ie procaine, lidocaine) is an attenuated effect following chronic cocaine administration. Thus, the effect of cocaine on cholinergic systems discussed below may be affected by non-dopaminergic processes, including a direct effect on ACh receptors.
Generalized Effects of the Cholinergic System on Reward and Drug Taking
Systematically administered cholinomimetics or cholinesterase inhibitors generally induce depressive-like behaviors, including learned helplessness. Antimuscarinic drugs reverse these behaviors and can produce euphorigenic-like effects (Janowsky and Overstreet, 2000; Lucki, 1997; Porsolt et al, 1978). Therefore, it might be predicted that compounds that increase cholinergic activity would decrease the rewarding effect of drugs. Consistent with this formulation were early observations that physostigmine, an acetylcholinesterase (AChE) inhibitor, decreases cocaine self-administration in rhesus monkeys (de la Garza and Johanson, 1982). On the other hand, rhesus monkeys trained to self-administer cocaine decreased their intake when administered atropine, an mAChR antagonist (Wilson and Schuster, 1973). Since physostigmine promotes ACh elevations both at mACh and nACh receptors, these studies may suggest that the mACh and nACh systems have opposing effects. In addition, physostigmine and atropine have significant generalized adverse effects that could effect responding. However, as detailed below, these inconsistencies persist when considering regionally specific alterations in mAChR systems in more recent, and far more extensive, preclinical studies.
VTA Acetylcholine and Reward
ACh agonists are rewarding when injected into the VTA. ACh infusions into the VTA potentiate ICSS (Redgrave and Horrell, 1976) and the ACh agonist carbachol and the AChE inhibitor neostigmine (which increases ACh concentrations) are both self-administered into the VTA (Ikemoto and Wise, 2002). M5 receptor-deficient (M5−/−) rats lack sustained NAc DA release following electrical stimulation of VTA cholinergic input (Forster et al, 2002), and striatal DA release induced by oxotremorine is significantly reduced in these M5 receptor-knockout mice (Zhang et al, 2002). The absence of M5 receptors on the VTA likely accounts for the lower rate of cocaine self-administration in M5−/− rodents (Fink-Jensen et al, 2003; Thomsen et al, 2005). Although α4–6β2 and α4–5β2 nAChRs have been identified on VTA DANs (Klink et al, 2001), VTA mACh receptors appear to play a more prominent role in maintaining reward relative to nAChRs. Using lateral hypothalamic brain stimulation as the reward, Yeomans and Baptista (1997) found that the intra-VTA administration of an mAChR antagonist (atropine) produced a rightward shift in the dose–response curve of self-stimulation (signaling slower rates of responding at a given dose) four-fold greater than that of two nAChR antagonists (mecamylamine and dihydro-beta-erythroidine (DHβE)). However, Smith et al (2004b) did not find a significant difference in VTA ACh concentrations in rats administered cocaine, either by self-administration or yoked, relative to yoked saline rats. Overall, the literature suggests that VTA ACh is rewarding, mediates the rewarding effects of cocaine and other positive reinforcers, and is primarily regulated by M5 receptor subtypes.
Striatal Acetylcholine and Reward
Striatal ACh increases following nonspecific, appetitive rewards. Mark et al (1992) reported an increase in extracellular NAc ACh following both feeding and water intake (following 20-h food or water deprivation, respectively). In a separate experiment, monitoring of NAc ACh at 10-min intervals during free-feeding revealed an increase in ACh immediately following maximal food intake (Mark et al, 1992). Rada et al (2005) also demonstrated a significant increase of ACh in the NAc shell following binging in ‘sucrose-dependent’ rats. Unlike the relatively quick increase in ACh observed by Mark et al (1992), Rada et al (2005) reported a maximal ACh after the sucrose meal ended. The involvement of NAc ACh in food reward appears to be mediated by mAChR, as the administration of scopolamine (1.0 or 10.0 μg/side) into the NAc core or shell inhibited both the appetitive learning and the number of lever presses expended for the sucrose, whereas mecamylamine (10.0 μg/side) did not (Pratt and Kelley, 2004). In a follow-up study, Pratt and Kelley (2005) reported that both ventrally and dorsally administered scopolamine into the striatum (0.5 or 10.0 μg/bilaterally) reduced 24-h food intake without affecting water intake.
Similar to elevations in VTA ACh concentrations following cocaine and striatal ACh concentrations following food intake, striatal elevations in ACh have also been associated with drug administration. Following a single dose of 10 and 20 mg/kg i.p. cocaine, for example, Imperato et al (1993a) found a 51 and 80% increase, respectively, in caudate nucleus ACh. Zocchi and Pert (1994) reported an increase in striatal ACh following 20 and 40 mg/kg i.p. cocaine, but not 10 mg/kg, and Mark et al (1999) also found an increase in shell NAc ACh in rats receiving yoked cocaine relative to those receiving saline. A similar finding in cocaine-infused rats, compared to saline-infused controls, was reported by Smith et al (2004b) in ACh turnover of the caudate putamen, but not the NAc. Additionally, Smith et al (2004b) found no significant differences in ACh concentrations of either region following 20 days of yoked cocaine infusion. Striatal increases in ACh following cocaine and d-amphetamine were blocked with the D1 antagonists SCH 39166 and SCH 23390 (Consolo et al, 1999; Imperato et al, 1993a), indicating that striatal increases in ACh were mediated by stimulant-induced increases in DA. In rats self-administering alcohol relative to yoked controls, Zocchi and Pert (1994) reported a similar dose-dependent increase in striatal ACh following morphine and a 51% increase in cyclin-dependent kinase 5 immunoreactivity, a measure of neuronal plasticity, in NAc shell cholinergic neurons (Camp et al, 2006). However, other investigators have found that the NAc dopaminergic efflux that follows the acute and/or chronic administration of ethanol (Rada et al, 2004), diazepam (Rada and Hoebel, 2005), and morphine (Rada et al, 1991) is not paralleled by an elevation in striatal ACh.
ACh striatal efflux has also been associated with the acquisition of cocaine. Mark et al (1999) found that rodents self-administering cocaine for 14 consecutive days significantly increased ACh release in the NAc shell more than rats non-contingently administered cocaine. Similarly, Crespo et al (2006) found that the run time (how fast an animal runs to obtain the contingent stimulus) was inversely correlated to ACh release in the NAc core during the acquisition of reminfentanil (a short-acting synthetic opioid) or cocaine (ie the shorter the run time, the greater the ACh release observed). Furthermore, acquisition of reminfentanil (cocaine was not tested) was blocked following the intra-accumbens administration of either scopolamine or mecamylamine (Crespo et al, 2006). Finally, Berlanga et al (2003) found a direct correlation (R2>0.90) between the percent of cholinergic neurons activated (as measured by Fos labeling) in the NAc shell and ventromedial striatum (but not the NAc shell or dorsolateral striatum) and the amount of cocaine self-administered in rats. Overall, these studies argue that cocaine, and possibly morphine and amphetamine, increases NAc ACh. Furthermore, the increase in ACh release appears to be positively correlated with the drug's positive reinforcing value. This striatal ACh elevation is likely a result of drug-induced D1 stimulation. Furthermore, the rewarding experience of cocaine acquisition is also associated with an elevation of striatal ACh.
The work of Smith et al offers a potentially contrasting view of the role of ACh in cocaine self-administration. Smith et al (2004b) found that the NAc ACh turnover did not differ between cocaine self-administering rats (or yoked saline-infused rats) and yoked cocaine-infused rats, and that caudate putamen ACh was significantly lower in the self-administering rats relative to the yoked cocaine-infused rats. The different directions of findings by Smith et al (2004b) may be related to their use of pulse labeling to assess ACh turnover, as turnover rate measurements for ACh require short pulse times. Their findings, therefore, represent only the turnover of ACh during the pulse interval, which would not have assessed ACh turnover during cocaine's more extended duration of action. Smith et al (2004a) also bilaterally ablated cholinergic neurons in the posterior-NAc-ventral palladium in rats trained to self-administer cocaine. Ablation was performed with 192-IgG-saporin; 192-IgG-saporin is directed to cell-surface antigens that are expressed at high levels only on cholinergic neurons. Interneuron destruction induced a shift in the dose–intake relationship to the left, such that lesioned animals self-administered cocaine at doses below the pre-lesion threshold in a dose-related manner. This work suggests that the removal of ACh interneurons (decreasing ACh efflux) increased the rewarding effects of cocaine, which would seem to contrast with the aforementioned literature positively associating heightened striatal ACh to cocaine acquisition. The complicated interplay of striatal ACh with other receptor systems, as well as the myriad effects of the multiple cholinergic subtypes (including presynaptic inhibitory and postsynaptic excitatory receptors), makes it difficult to predict a specific direction of change in response to a generally destructive agent or a nonspecific agonist or antagonist.
Unlike ACh increases in VTA, however, there appears to be little evidence that striatal ACh is itself rewarding. In fact, striatal ACh is often associated with aversion. Aversive-related increases in striatal ACh are observed in response to the injection of a vehicle (Pfister et al, 1994), the conditioned stimulus of an aversive taste (Mark et al, 1995), and following inescapable automatic hypothalamic stimulation (Rada and Hoebel, 2001). When allowed to escape from the hypothalamic stimulation, extracellular ACh concentrations are significantly decreased (Rada and Hoebel, 2001). High levels of NAc ACh persist for up to 24 h following the Porsolt, or forced swimming, test following an early transient decrease (Rada et al, 2006). (The Porsolt test is a measure of ‘behavioral despair,’ or learned helplessness.) Microinjections of arecholine, a cholinergic agonist, into the NAc dose-dependently inhibit swimming in the Porsolt in rodents, and pirenzepine, an M1 antagonist, increases swimming (Chau et al, 2001). Thus, increases in striatal ACh activation appear to heighten learned helplessness. Gallamine, an M2 antagonist, also decreases swimming, presumably by blocking ACh autoreceptors and thereby increasing ACh concentrations (Chau et al, 2001). Hoebel and colleagues have also noted a dramatic increase in striatal ACh, relative to DA, concentrations during naloxone-induced morphine withdrawal (Rada et al, 1991), mecamylamine-induced nicotine withdrawal (Rada et al, 2001), flumazenil-induced diazepam withdrawal (Rada and Hoebel, 2005), naloxone-induced ethanol withdrawal (Rada et al, 2004), and following the presentation of a conditioned aversive stimulus in a taste aversion task (Mark et al, 1991, 1995). Electrophysiologic studies further reveal that the response of the cholinergic TANs differs following aversive stimuli relative to reward (Apicella, 2007).
Thus, there is now a substantial literature demonstrating a role of ACh in the acute administration of cocaine and the interpretation of its reinforcing effect. It remains unclear, however, exactly how ACh alters the perception of cocaine reinforcement. Rada and Hoebel (2000) have suggested a specific mechanism whereby NAc ACh has a satiating effect upon appetitive stimulus which complements the facilitatory effects of NAc DA. The interactive effects of DA and ACh then mutually contribute to produce a modulatory effect on other receptor systems. While there is evidence to support this interpretative, much of the above-referenced literature would be difficult to reconcile with this model.
Finally, the above section has used the term ‘striatal’ to describe a complex system comprised of multiple distinct regions with quite different functions. Although specific regions (ie NAc shell or core, caudate, putamen, dorsal striatum) were often noted, the referenced literature frequently did not isolate a more demarcated region. Given the different prescribed roles of the dorsal and ventral striatum (Gerdeman et al, 2003; White, 1996), it is presumed that the location of the ACh interneurons may have a significant effect on interpretation—although no such relationship was apparent in the reviewed studies. It is also noteworthy that the dorsal/ventral striatal distinction has recently come under question (suggesting a ventromedial vs dorsolateral distinction) (Voorn et al, 2004), further complicating any attempt to ascribe regional effects of striatal ACh. As noted previously, the excitatory and inhibitory effects of striatal ACh further complicate any generalized assumption of cholinergic effect and cocaine administration.
NEUROADAPTATION
ACh Receptor Alterations Following Cocaine
Persistent elevations in striatal ACh might be expected to produce a compensatory downregulation of striatal ACh receptors. Decreases in both mACh receptor density and Kd were, in fact, observed by Macedo et al (2004) following 7 continuous days of cocaine administration. Decreases in receptor density and dissociation constants were also identified with [3H]-N-methylscopolamine at 30 min, 5 days, and 30 days following the cessation of cocaine administration (20 and 30 mg/kg). Zeigler et al (1991) also found a decrease of mACh receptors, as measured by quinuclidinyl benzilate binding, in both NAc and medial caudate regions following the implantation of a 5-day, slow-release pellet. Interestingly, a matched group of rats administered the same amount of cocaine over 5 days by daily, i.p. injections did not demonstrate similar reductions, suggesting that the rate of cocaine administration (continuous vs daily) significantly affects receptor disruption. Wilson et al (1994) reported a 26% reduction in mean NAc choline acetyltransferase (ChAT) (an enzyme that controls the production of ACh) activity in rat NAc immediately following the chronic, unlimited access to cocaine (approximately 90 mg/kg/day) (NAc (−32%), striatum (−18%)). This reduction continued for 3 weeks (49±5 days) (the last time point measured), suggesting a persistent compensatory decrease in ChAT in response to cocaine-induced elevations in striatal ACh. In contrast, Sousa et al (1999) reported an increase in both M1-like (assessed with [3H]-N-methylscopolamine in the presence of carbachol) and M2-like (assessed with [3H]-N-methylscopolamine in the presence of pirenzepine) mACh receptors following 7 days of cocaine administration (5 and 10 mg/kg), albeit at a lower dose of cocaine than that used by Macedo et al (2004). Finally, Lipton et al (1995) observed both an increase and a decrease in striatal mACh (assessed with [3H]-quinuclidinyl benzilate) receptors following 5 days of cocaine administration, depending on whether measures were obtained 12 h, 2 days, or 21 days after the cessation of cocaine injections. In toto, these studies suggest a downregulation of the cholinergic system following cocaine administration and are consistent with an adaptive response to increased striatal cholinergic release during cocaine administration.
However, this literature is somewhat ambiguous, mirroring the more extensive literature assessing repeated cocaine administration on striatal D1 and D2 receptor density. Studies have variously reported receptor downregulation (Maggos et al, 1998; Zeigler et al, 1991), upregulation (Alburges et al, 1992, 1993; Macedo et al, 2004), and no alterations (Mayfield et al, 1992; Sousa et al, 1999) in D1 densities between days 1 and 30 of withdrawal. Similar inconsistencies are reported with respect to D2 receptors (upregulation (Macedo et al, 2004; Sousa et al, 1999; Zeigler et al, 1991); downregulation (Maggos et al, 1998; Volkow et al, 1993); no change (Alburges et al, 1992, 1993; Maggos et al, 1998)). Given the complex interplay between mesostriatal ACh and DA described earlier, it is perhaps not surprising that the extant literature is inconsistent with respect to the direction of striatal cholinergic change resulting from, or effecting, cocaine administration.
Cocaine-Induced Sensitization
Cocaine-induced sensitization in the VTA. Behavioral sensitization refers to the progressive increase in the locomotor stimulant properties of a drug following chronic drug administration and is considered a long-term neuroadaptation important for the maintenance of drug dependence (Kalivas et al, 1998; Robinson and Berridge, 1993; Vanderschuren et al, 1999). Cocaine-induced sensitization occurs through the stimulation of D1 receptors on cortical afferents to the VTA, which, in turn, induces glutamate release. Glutamate activates ionotropic glutamate receptors located on DA neurons, triggering the long-term molecular adaptations that underlie sensitization (Kalivas, 1995). nAChRs are expressed on VTA neurons as well as cortical glutamate afferents to the VTA; β2-containing nAChRs are expressed on DA and GABAergic neurons while α7 nAChRs are primarily expressed on glutamatergic terminals (Klink et al, 2001). Both the heightened dopaminergic response and the psychomotor effects of cocaine-induced sensitization are prevented by mecamylamine, an nAChR antagonist that blocks both α7- and β2-containing VTA nAChRs (Schoffelmeer et al, 2002). Champtiaux et al (2006) have subsequently identified the VTA heteromeric β2-containing nAChRs, but not homomeric α7 nAChRs, as critical for the induction of cocaine-induced locomotor sensitization; intra-VTA microinjections of the α4β2-selective containing antagonist DHβE, but not the α7 antagonist methyllycaconitine, prevented the induction of behavioral sensitization to cocaine. On the other hand, Zanetti et al (2006) found that whereas the co-administration of DHβE and methyllycaconitine prevented sensitization to cocaine (as measured by extracellular DA release in the ventral striatum), neither antagonist was effective in preventing sensitization alone. Although differences in frequency and amount of cocaine dose and the interval following cocaine administration at which sensitization was assessed differed between the two studies, the reason for these distinctly different results is not readily apparent.
Cocaine-induced sensitization in the NAc. NAc mAChRs also exert an inhibitory influence on cocaine-induced behavioral sensitization (Wolf, 1998). Although Itzhak and Martin (2000) found no effect of scopolamine (1.0 mg/kg s.c.) on cocaine (20 mg/kg i.p.)-induced ‘place-independent’ sensitization, Hikida et al (2001) examined the role of accumbens ACh in cocaine-induced sensitization by ablating these interneurons with an immunotoxin-mediated cell targeting technique. Transgenic mice with bilateral ACh interneuron ablation displayed a prominent and progressive increase in locomotor activity at baseline and following daily cocaine administration (5, 10, and 20 mg/kg) relative to their wild-type littermates. Using the same technology, Hikida et al (2003) also reported that administration of the AChE inhibitor donepezil (which increases ACh synaptic concentrations) prior to cocaine administration (10 mg/kg, 3 days) blocked induction of locomotion sensitization to cocaine in rats. Furthermore, the AChE inhibitors donepezil and galanthamine also blocked hyperlocomotion after the establishment of locomotor sensitization (5 days, 10 mg/kg). This inhibition was abolished by ablation of the NAc cholinergic interneurons. Although the preceding studies suggest that ACh inhibits sensitization, Heidbreder and Shippenberg (1996) found that subcutaneously administered scopolamine blocked the induction, but not the expression, of cocaine-induced sensitization (20 mg/kg i.p., 5 days). Microinjections of the nicotinic antagonist DHβE into the NAc had no effect on cocaine-induced sensitized locomotor activity (Champtiaux et al, 2006).
Although mAChRs play an important role in VTA reward mediation, it appears that nAChR receptors are primarily involved in cocaine-induced behavioral sensitization in the VTA. In contrast, ACh exerts an effect on NAc cocaine-induced sensitization through mAChRs, although the direction of this effect is uncertain. As discussed below, striatal cholinergic activity also appears to heighten context-dependent locomotor sensitization.
ACETYLCHOLINE, LEARNING, AND MEMORY
Learning and memory are essential to the changes in behavior produced by addictive drugs. Drugs reinforce associations that initiate the storage of new information, strengthening neural pathways through synaptic plasticity and long-term potentiation (Calabresi et al, 2000; Everitt and Robbins, 2005; Hyman et al, 2006; Suzuki et al, 2001). Acetylcholine is involved in both explicit and implicit memories, utilizing distinct neuronal systems.
Acetylcholine and Explicit Memory
Explicit memories (also called declarative memory) refer to the learning and memory of facts and events that require conscious recall (Tulving and Schacter, 1990). Explicit learning occurs within conscious awareness and with relatively little experience (Breese et al, 1989; Sharp et al, 1985). Through explicit memories related to drug use (ie, where the substance was obtained; where it was used; the social situation), as well as memories of the effects of the drug on internal affective states, the drug-addicted individual learns to manipulate the external environment to achieve desired affective states (Berke and Eichenbaum, 2001; Hirsh et al, 1979). Thus, this type of learning enables an individual to evaluate the outcome of their actions and to modify their future behavior accordingly, including either the induction of a hedonic state or the alleviation of an unpleasant affective state.
The locus of explicit memory is the hippocampus. The hippocampus contains among the highest concentrations of ACh in the CNS, and ACh is a critical component of hippocampal memory formation (Grecksch et al, 1978; Jaffard et al, 1980; Singh et al, 1974). Hippocampal ACh is projected from the septum and nucleus of the diagonal band (McKinney et al, 1983), and DA transmission at D1 and/or D2 receptors can enhance hippocampal ACh output (Imperato et al, 1993b). Drugs that block mAChRs, such as scopolamine, impair the encoding of new information (Hasselmo and Fehlau, 2001; Hasselmo et al, 2002) and depletion of ACh is linked to memory disorders such as Alzheimer's disease (Coyle et al, 1983; Quirion et al, 1989). Hippocampal ACh is theorized to facilitate the encoding of new information by simultaneously suppressing excitatory synaptic transmission and enhancing long-term potentiation, while leaving excitatory feed-forward synapses relatively unaffected (Hasselmo et al, 2002). ACh also modulates synaptic plasticity within the hippocampus (Colgin et al, 2003) and long-term potentiation is drastically reduced in genetically engineered mice lacking the mACh M2 receptor (Seeger et al, 2004).
The acute administration of cocaine increases extracellular levels of ACh (Imperato et al, 1993a, 1996) and ACh turnover rates within the hippocampus (Robinson and Hambrecht, 1988). Enhancement of hippocampal ACh release induced by cocaine appears to occur through DA D1 receptor activation, as ACh release was fully antagonized by the D1 antagonist SCH 23390 (Imperato et al, 1993a; Robinson and Hambrecht, 1988). Cocaine-induced increases in hippocampal ACh are consistent with the downregulation of mACh receptor binding observed by Zeigler et al (1991) in rats continuously administered cocaine for 5 days (paradigm described earlier). On the other hand, Smith et al (2004b) found that a cocaine-yoked group of rats showed lower, not higher, concentrations of hippocampal ACh relative to a saline-yoked group. However, rats self-administering cocaine exhibited a significant elevation in both hippocampal ACh levels and ACh turnover relative to the cocaine-yoked group (Smith et al, 2004b), suggesting that the process of self-administering cocaine, and not the cocaine itself, is the critical component in increasing hippocampal ACh concentrations. Although these studies, in toto, are inconsistent with respect to the directional relationship between cocaine administration and hippocampal ACh release, they strongly suggest that changes in hippocampal ACh during investigator- and self-administration of cocaine may be involved in the solidification of hippocampal-mediated cocaine-related explicit memories. To our knowledge, however, this relationship has not been empirically confirmed.
Acetylcholine and Conditioned Learning
Implicit memory (also called nondeclarative memory) refers to the learning of perceptual and/or motor skills that usually follows repetition. These memories become reflexive once associations are formed, operating largely outside of awareness (Roediger, 1990; Schacter, 1992). Conditioned learning (also called associative learning), a type of implicit learning, is based on the principals of classical conditioning (Pavlov, 1927). Through this process, neutral stimuli acquire reinforcing qualities and motivational relevance, even in the absence of a drug. Since implicit memories are difficult to extinguish, they appear central to both the maintenance of cocaine addiction and relapse (Everitt and Robbins, 2005; White, 1996). This process forms the basis of drug-induced CPP and, relative to other types of conditioned learning, appears more difficult to extinguish. As there is an absence of habituation to drug-induced increases in DA (relative to natural rewards), the associations between the rewarding properties of cocaine and the related conditioned stimuli are continually strengthened with repeated use (Di Chiara et al, 2004). The NAc, amygdala, and basal forebrain all play a critical role in the development of conditioned learning following drug administration (Di Chiara et al, 1999; Everitt et al, 1999; Meil and See, 1997).
Conditioned learning and the VTA. CPP paradigms are most often used to assess the strength of associated stimuli with drug administration. The ACh agonist carbachol (Yeomans et al, 1985) and the nicotinic agonist cystisine (Museo and Wise, 1994) induce CPP when injected into the VTA. Conversely, lesions of the pedunculopontine nucleus (a source of ACh to the VTA) inhibit the development of CPP to morphine and amphetamine (Bechara and van der Kooy, 1989; Olmstead and Franklin, 1993). M5−/− rodents also spend less time in a cocaine-paired compartment during a CPP procedure compared to wild-type rodents (Fink-Jensen et al, 2003). Consistent with the positive association of VTA ACh with reward, increasing both muscarinic and nicotinic cholinergic VTA activity appears to heighten CPP to drugs of abuse.
Conditioned learning and the striatum. Although ablation of cholinergic striatal neurons did not impair either contextual or cued fear conditioning in rats (Kitabatake et al, 2003), infusions of scopolamine into the dorsal striatum impaired the conditioned learning of a radial arm task when administered soon after training (Legault et al, 2006). With respect to cocaine, ablation of NAc cholinergic interneurons by an immunotoxin-mediated cell targeting technique induced CPP for cocaine at much lower doses of cocaine than that in wild-type littermates (Hikida et al, 2001). The administration of AChE inhibitors (which increase ACh synaptic concentrations) also suppressed cocaine-induced CPP, and this effect was blocked by the ablation of ACh cells within the NAc (Hikida et al, 2003). These findings suggest that NAc ACh interneurons inhibit the associative learning that underlies cocaine addiction.
The association of a novel environment to stimulant administration has a profound effect on the development of behavioral sensitization (Browman et al, 1998; Crombag et al, 1996; Robinson et al, 1998). Seemingly in contrast to the findings described with CPP paradigms, Itzhak and Martin (2000) found that cocaine-induced place-dependent sensitization (context dependent), as measured by locomotor activity, was blocked by pretreatment with scopolamine (1 mg/kg s.c.). In this study, cocaine alone was administered for 5 consecutive days and produced a five-fold increase in locomotor activity. The administration of scopolamine 30 min prior to each cocaine injection completely abolished this increase. This is similar to findings described earlier, in which scopolamine (3 mg/kg s.c.) was observed to block the induction, but not the expression, of context-independent sensitized locomotor activity (Heidbreder and Shippenberg, 1996). However, Itzhak and Martin (2000) did not observe a disruptive effect of scopolamine on the induction of context-independent sensitization by cocaine, possibly due to the lower dose of scopolamine. Although these studies involve peripherally administered cholinergic agents, the association of mesostriatal DA with sensitization would be consistent with a cholinergic striatal mechanism of action.
Thus, increased striatal cholinergic activity appears to inhibit the development of cocaine-induced CPP but heighten context-dependent locomotor sensitization. The former findings are puzzling and somewhat counterintuitive. Striatal ACh is a critical component in the switch from acquisition to consolidation (Rasch et al, 2006). It would, therefore, be expected that an increase in striatal ACh would be associated with not only the increased salience of cocaine's effect (as described in a previous section), but also the salience of associated stimuli. The absence of this effect (in fact, the reverse is observed: increased striatal ACh attenuated (Hikida et al, 2003) and decreased striatal ACh augmented (Hikida et al, 2001) cocaine-induced CPP) may, in part, be due to the NAc target of these studies, as conditioned learning is primarily a function of the dorsal striatum. In particular, the association of TANs with associative learning has been confined to the dorsal striatum (Apicella, 2007). Related findings in the amygdala (below) are also directly relevant.
Conditioned learning and the amygdala. The basolateral portion of the amygdala facilitates the processing of stimuli that are behaviorally relevant when stimulus–outcome associations are being formed (van der Zee and Luiten, 1999). Through this process, environmental cues paired with drug use acquire motivational salience valiance and guide goal-directed responses to these stimuli (See, 2005). Conditioned learning is mediated by basolateral amygdala (BLA) mAChRs, at least in part, as the bilateral infusion of the non-selective mAChR agonist oxotremorine into this region significantly improved retention of an inhibitory avoidance task (Power et al, 2003). Also, infusions of the mAChR agonist scopolamine into the BLA impaired the learning of a food CPP task (McIntyre et al, 1998). Both M1 and M2 ACh receptors appear to be involved, as both telenzipine (a selective M1 antagonist) and methoctramine (a selective M2 antagonist) blocked the retention of the avoidance task described above (Power et al, 2003).
The amygdala is one of the few brain regions that consistently exhibit increased metabolic activity in response to cocaine-associated cues in both animal (Brown et al, 1992; Ciccocioppo et al, 2001) and human (Childress et al, 1999; Kilts et al, 2001) studies, consistent with this region's role in conditioned emotional memories. In preclinical studies, the infusion of scopolamine into the BLA just prior to the acquisition phase of a classical conditioning procedure produced a dose-dependent disruption of cocaine-seeking behavior during cue-induced drug reinstatement (See et al, 2003). Drug reinstatement was unaffected when scopolamine was administered in the BLA following successful conditioning and just prior to the expression of conditioned-cue reinstatement (See et al, 2003). This is consistent with the disruption of a CPP-related food task with scopolamine described above (Power et al, 2003). Interestingly, Zeigler et al (1991) have shown a decreased number of amygdalar mACh receptors following the administration of cocaine over 5 days by an implanted pellet (paradigm described earlier). Thus, BLA mAChRs appear to play a role in the acquisition of cocaine-related associations, but not in the expression of previously acquired associations. In contrast, intra-amygdalar injections of the mACh agonist oxotremorine (10 ng/0.5 μl) also facilitated the extinction of amphetamine (2 mg/kg) CPP when administered immediately following the training trial, but not if administered 2 h after the training trial (Schroeder and Packard, 2004). Although both the See et al (2003) and Schroeder and Packard (2004) studies reveal that cholinergic interventions suppress the acquisition of stimulant-related associations, the direction of change is different; See et al (2003) showed disruption with a cholinergic antagonist, whereas Schroeder and Packard (2004) used a cholinergic agonist. These differences may be a result of the drug association used (cocaine vs amphetamine, respectively) and/or the behavior assessed (acquisition vs extinction, respectively).
ACETYLCHOLINE AND PREFRONTAL CORTICAL COGNITION
Executive cognitive mechanisms provide the behavioral flexibility to override automatic responses that are no longer adaptive. Examples of executive processes include attentional selection and resistance to interference, behavioral inhibition, set shifting, planning, and decision making. Executive processes primarily occur in the PFC. Acetylcholine has been implicated in several aspects of executive function that appear to be impaired in cocaine addicts, most notable being attentional processes, response inhibition, and set shifting (Adinoff et al, in press; Horner et al, 1996; Rilling and Adinoff, 2007).
Attentional processes. Attention refers to the different capacities or processes that relate to how an organism becomes receptive to stimuli and how it begins to process incoming or attended-to (internal or external) excitation (Parasuraman, 1998). ACh is strongly linked to these attentional processes (Baxter and Chiba, 1999; Robbins, 2002; Sarter et al, 2003). ACh efflux, for example, is increased into the medial frontal cortex during a five-choice serial reaction time task (5-CSRTT)—a measure of visual attention. Rats receiving highly selective ACh immunotoxins (192 IgG-saprin) into the NBM (which projects to the PFC and amygdala) display dose-related decrements in attention parameters on a 5-CSRTT (McGaughy et al, 2002). Specifically, the number of ChAT-immunoreactive cells in NBM was significantly correlated with task accuracy (McGaughy et al, 2002). The effects of similar basal forebrain lesions (induced by quisqualate) on a visual attention task were reversed following the implantation of ACh-enriched neural grafts into the cortex or following systemic treatment with physostigmine or nicotine, supporting the relevance of ACh to this process (Muir et al, 1992, 1995). The rise in NAc DA resulting from exposure to a salient stimulus may focus attention by stimulating cortical ACh efflux (Acquas et al, 1994; Day and Fibiger, 1992, 1994; Sarter et al, 1999). As noted for striatal and hippocampal increases in ACh, cortical elevations in ACh are blocked by D1 antagonists (Acquas et al, 1994).
Long-term cocaine abuse has been linked to attention and concentration deficits on standardized neurocognitive measures (Ardila et al, 1991; Horner et al, 1996; Jovanovski et al, 2005). In a review of 17 studies, Horner et al (1996) tentatively concluded that chronic cocaine use appears to decrease cognitive speed. A more recent review of 15 studies by Jovanovski et al (2005) suggested significant deficits in cocaine-addicted subjects on measures that required sustained, focused, and divided attention. In preclinical studies, Dalley et al (2005) pre-trained rats in the 5-CSRTT and then exposed them to multiple ‘long-access’ cycles (five daily sessions repeated on four successive occasions) of cocaine self-administration. Impaired attentional accuracy, increased omissions, and slower latencies were observed 24 h, but not 1 month, following the cessation of cocaine administration. There was, however, no effect on visual attentional performance relative to surgically controlled rats. To our knowledge, there are no empirical studies directly connecting cocaine-induced deficits in attention with ACh.
Set shifting. Set shifting, or response reversal, is a key element of decision making and is required when response contingencies, such as the amount of reward, direction of reward (win or lose), or the time it takes to obtain a reward, are altered. The dorsomedial striatum is one of the regions critical for this behavioral flexibility, and recent experiments suggest that the activation of ACh interneurons within the dorsomedial striatum facilitates behavioral flexibility under conditions of changing contingencies (Ragozzino and Choi, 2004; Ragozzino et al, 2002). Activation of muscarinic cholinergic receptors in this region may facilitate the learning of situationally adaptive response patterns. For example, ACh efflux in the dorsal striatum did not change during the acquisition phase of a discrimination task, but increased during the reversal stage as a rat was beginning to learn a new response pattern (Ragozzino and Choi, 2004). Striatal ACh then returned to baseline after a new response pattern was reliably executed. Infusion of the mAChR antagonist scopolamine (8 μg/side, but not 1 μg/side) into the dorsomedial striatum did not impair learning in a simple response discrimination task, but did impair response reversal learning (Ragozzino et al, 2002). Chen et al (2004) also found that the systemic administration of scopolamine (0.1 and 0.25 mg/kg i.p.) dose-dependently blocked learning of set-shifting paradigms. Although the systemic administration of scopolamine did not allow regional localization of this effect, the absence of an effect by methylscopolamine (which does not cross the blood–brain barrier) demonstrated central mechanisms (Chen et al, 2004). Palencia and Ragozzino (2006) also observed an increase in dorsomedial striatal efflux during reversal learning in rats. These investigators suggested that this ACh effect may, at least in part, be mediated by N-methyl-D-aspartate (NMDA) glutamate receptors, as dorsomedial NMDA receptor blockade by dl-2-amino-5-phosphonopentanoic acid significantly impaired both response reversal learning and concomitant ACh efflux.
The addictive use of cocaine has been linked to poor performance on set-shifting tasks, which require learning new strategies and reject old strategies that are no longer effective (Adinoff et al (in press); Rilling and Adinoff, 2007). Impaired performances on such response reversal measures such as the Wisconsin card sorting task (WCST) (Ardila et al, 1991; Beatty et al, 1995; Rosselli et al, 2001) and the trail making test (Trails B) (Beatty et al, 1995; Rosselli et al, 2001; Smelson et al, 1999; Strickland et al, 1993) have been demonstrated in cocaine-addicted subjects relative to controls. Although the preclinical literature does not presently provide a link between set-shifting deficits following cocaine administration and alterations in cholinergic systems, recent work from our clinical laboratory suggests such a connection (B Adinoff et al, unpublished). In these studies, physostigmine and saline were administered to abstinent cocaine-addicted subjects and healthy controls while at rest. Regional cerebral blood flow (rCBF), a measure of neural activity, was assessed with single photon emission computed tomography (SPECT) following each infusion. On a third study day, set shifting was assessed with both the WCST and intradimensional/extradimensional shift (IED) task (Cambridge, 2002). Scores on each task were then correlated with rCBF following physostigmine (relative to saline) in the orbitofrontal cortex (OFC), a region critical for set-shifting processing. A significant correlation was observed between OFC rCBF following physostigmine and both WCST and IED scores in the healthy controls, but not in the cocaine-addicted subjects. These findings offer preliminary evidence that cholinergic disruption following chronic cocaine use may be associated with deficits in decision making.
SUMMARY AND FUTURE DIRECTIONS FOR CLINICAL RESEARCH
Given the multiple origins and discrete projections of CNS cholinergic neurons and their involvement in a myriad of cognitive functions, the relationship between cocaine and ACh is predictably complicated. Acutely, cocaine administration increases ACh in the VTA, NAc, and dorsal striatum. Elevations in VTA ACh concentrations following acute cocaine occur via neuronal projections from the mesopontine nuclei and heighten cocaine's rewarding effects, primarily through the M5 receptor, by complementing DA neuronal input in a feed-forward manner. In contrast, NAc and dorsal striatal increases in ACh are produced via VTA DA afferents upon striatal ACh interneurons. Striatal ACh release (under the modulating influence of D1 and D2 receptors) appears to increase cocaine's rewarding effects but decrease cocaine-associated conditioned learning. The chronic administration of cocaine also appears to result in fewer ACh receptors upon abstinence, although the direction of these changes is dependent on cocaine dose, duration, time since last administration, and brain region assessed. nAChRs facilitate the initiation of VTA cocaine-induced sensitization, whereas NAc mAChRs inhibit NAc sensitization.
Cocaine reinforces different types of learning by initiating the storage of new information and strengthening neural pathways through the process of synaptic plasticity. Since cocaine also increases extracellular levels of ACh and ACh turnover rates within the hippocampus, high levels of hippocampal ACh during cocaine use may facilitate the encoding of explicit drug-related information. The acquisition of cocaine-related associations thought to underlie craving is altered by cholinergic release in both the amygdala and striatum, although the direction of change effected is uncertain. Although ACh likely plays a role in the cocaine-related deficits observed in executive functioning, this relationship has yet to be demonstrated and remains a critical area of investigation.
In summary, a plethora of studies now clearly document that the cholinergic system is involved to a great extent in both the experience of cocaine and the conditioned association of cocaine with salient stimuli. This association is complicated by the multiple origins of cholinergic neurons, the unique aspects of striatal cholinergic neurons, and the presence of inhibitory and excitatory ACh subtypes. Our review was also focused on mACh receptor systems with only limited attention on the nACh receptors. Internal and external feedback systems, specific for each cholinergic system, further complicate interpretation. Thus, clarification of the role of ACh in cocaine addiction requires increasing use of agonists and antagonists specific for receptor subtype, as well as animals bred with specific subtype deficits.
The above-reviewed studies do not involve humans, with the exception of a few. These preclinical studies, however, strongly support a role of ACh receptor systems in the progression of cocaine from initial reward to the maintenance of addictive-like behaviors. Complementary clinical laboratory studies to examine the link between chronic cocaine use and ACh receptor alterations, as well as pharmacological trials to assess the utility of cholinomimetics in the treatment of cocaine dependence, are now required. Initially, neuroimaging studies can be used to investigate alterations in ACh receptor systems in cocaine-addicted subjects. SPECT, PET, and functional magnetic resonance imaging (fMRI) techniques have been used to great advantage in the investigation of cholinergic functioning in other populations, particularly in Alzheimer's disease (Volkow et al, 2001). For example, PET and SPECT ligands are available to assess extracellular ACh, ACh receptors, and brain function (ie brain blood flow and glucose utilization) as well as markers of cholinergic cell viability (vesicular transporters, AChE) (Zubieta et al, 1998). Radioligands can be used to identify mAChRs (Volkow et al, 2001) and nAChRs (Ma et al, 2002), and more recently developed tracers can select for M2 receptor subtypes (Furey et al, 2000). These techniques may also be used to explore cocaine-related deficits in executive function, such as learning tasks relevant to the addictive process, in the presence or absence of cholinergic agonist or antagonists. For example, Furey et al (2000) have utilized fMRI to explore cognitive-induced alterations in neural activation during a working memory task concurrent with the infusion of physostigmine. Given the importance of ACh in modulating the tonic and clonic release of DA, the concurrent functional assessment of DA and ACh systems during acute cocaine administration, withdrawal, and extended abstinence in addicted patients may elucidate the convergent interaction and relevant disruptions in these two complementary systems.
In the first, and as yet preliminary, study to utilize neuroimaging techniques to assess ACh systems in cocaine-addicted subjects, we have used SPECT to identify alterations in ACh systems in abstinent cocaine-addicted subjects. Preliminary findings suggest regional-specific changes in cerebral blood flow following the infusion of physostigmine or scopolamine compared to saline (Adinoff et al, 2005). To date, the only published studies exploring neural cholinergic systems in this population have reported that ChAT and vesicular ACh transporter (VAChT) concentrations in the autopsied brains of cocaine-addicted subjects did not differ from those of heroin-addicted (VAChT, ChAT) or control subjects (VAChT) (Kish et al, 1999; Siegal et al, 2004).
Ultimately, pharmacologic investigations will be required to explore the relevance of ACh alterations to the addictive process. Although cocaine increases ACh during acute administration, the literature reviewed above, in toto, suggests that ACh agonists may be the most promising agents in the treatment of cocaine dependence. Cholinomimetics may compensate for the apparent reduction in ACh receptors observed during withdrawal and facilitate the acquisition of non-cocaine-driven behaviors by strengthening the salience of stimuli unassociated with cocaine use. A large number of cholinergic agonists are now available for human use, and pharmaceutical agents that target specific receptor subtypes are rapidly being developed. Some of the most common cholinergic agonists are cholinesterase inhibitors, and donepezil, galantamine, and rivastigmine are presently available for use in humans (Cummings, 2000; Ellis, 2005). In a preliminary trial of donepezil using the Cocaine Rapid Efficacy and Safety Trial (CREST) study design, however, dozepezil did not produce significant changes in cocaine use (Winhusen et al, 2005). Treatment trials of rivastigmine in the treatment of cocaine addiction are ongoing. The relevance of the M5 receptor in cocaine reinforcement (Fink-Jensen et al, 2003; Thomsen et al, 2005) suggests that targeting this muscarinic subtype may be of particular importance.
In contrast, a clinical laboratory study revealed that the nicotinic receptor antagonist mecamylamine decreased craving in cocaine-dependent subjects (Reid et al, 1999), although its effectiveness in relapse prevention is unknown. This mirrors preclinical work demonstrating that mecamylamine suppressed both nicotine and cocaine self-administration (Blokhina et al, 2005). These studies highlight the difficulty of dissembling the contributions of cocaine vs nicotine to nAChR changes in cocaine- (and typically nicotine-) addicted individuals. In preclinical studies, the administration of nicotine alters the locomotor effects of cocaine (Collins and Izenwasser, 2004), the two drugs exhibit cross-tolerance (Desai and Terry, 2003), nicotine substitutes for cocaine reinforcement (Tessari et al, 1995), and repeated nicotine exposure enhances cocaine-seeking behavior (Bechtholt and Mark, 2002). In humans, nicotine increases cue-induced cocaine craving (Reid et al, 1998). Thus, the frequent co-morbid dependence on nicotine and cocaine, their shared effects on reward systems (Kauer, 2003), and their apparent interactive effects on the ACh system suggest that the nAChR may be a fruitful area of investigation. The newly available (for nicotine dependence) partial α4β2 nicotinic agonist, varenicline, may therefore be worthy of investigation for the treatment of cocaine dependence.
Perhaps one of the most promising treatment strategies involves the shared role of both DA and ACh in regulating reward and each other's synaptic release. From a clinical perspective, this neurobiologic interconnectiveness suggests that a combination of cholinergic and dopaminergic modulation may be the optimal treatment approach for the cocaine-dependent patient. For example, most studies of DA agonists have not provided strong signals in the treatment of cocaine dependence (Adinoff, 2004). The addition of a cholinomimetic may enhance the effects of a dopaminergic agonist in the NAc. Alternatively, a pharmacologic cocktail of DA agonist and cholinergic antagonist may provide the optimal environment during the early phase of cocaine abstinence. Clearly, other neurotransmitter systems that regulate ACh outflow and pharmacogenetic influences are also potent areas of investigation.
Ultimately, one of the greatest benefits derived from ACh modulation in the treatment of cocaine addiction may stem from its effects on learning and memory processes. These changes may account for some of the more intractable aspects of cocaine addiction, such as craving, sensitization, and conditioned learning. A better delineation of the molecular and cellular changes that occur during chronic cocaine use may provide important insights into the role of ACh in cocaine addiction, assisting in the development of stage- and site-specific pharmacological treatment interventions.
References
Acquas E, Day JC, Fibiger HC (1994). The potent and selective dopamine D1 receptor agonist A-77636 increases cortical and hippocampal acetylcholine release in the rat. Eur J Pharmacol 260: 85–87.
Adinoff B (2004). Neurobiologic processes in drug reward and addiction. Harv Rev Psychiatry 12: 305–320.
Adinoff B, Devous MD, Best SM, George MS, Alexander D, Payne K (2001). Limbic responsiveness to procaine in cocaine-addicted subjects. Am J Psychiatry 158: 390–398.
Adinoff B, Rilling LM, Williams MJ, Schreffler ER, Schepis TY, Rosvall T et al (in press). Impulsivity, neural deficits, and the addictions: the ‘Oops’ factor in relapse. J Addict Dis.
Adinoff B, Williams MJ, Best SE, Zielinski TA, Harris TS, Schreffler ER et al (2005). Cholinergic receptor systems in cocaine-addicted subjects: alterations in regional cerebral blood flow. Neuropsychopharmacology 30: S84.
Alburges ME, Hunt ME, McQuade RD, Wamsley JK (1992). D1-receptor antagonists: comparison of [3H]SCH39166 to [3H]SCH23390. J Chem Neuroanat 5: 357–366.
Alburges ME, Narang N, Wamsley JK (1993). Alterations in the dopaminergic receptor system after chronic administration of cocaine. Synapse 14: 314–323.
Alcantara AA, Chen V, Herring BE, Mendenhall JM, Berlanga ML (2003). Localization of dopamine D2 receptors on cholinergic interneurons of the dorsal striatum and nucleus accumbens of the rat. Brain Res 986: 22–29.
Aosaki T, Tsubokawa H, Ishida A, Watanabe K, Graybiel AM, Kimura M (1994). Responses of tonically active neurons in the primate's striatum undergo systematic changes during behavioral sensorimotor conditioning. J Neurosci 14: 3969–3984.
Apicella P (2002). Tonically active neurons in the primate striatum and their role in the processing of information about motivationally relevant events. Eur J Neurosci 16: 2017–2026.
Apicella P (2007). Leading tonically active neurons of the striatum from reward detection to context recognition. Trends Neurosci 30: 299–306.
Apicella P, Legallet E, Trouche E (1997). Responses of tonically discharging neurons in the monkey striatum to primary rewards delivered during different behavioral states. Exp Brain Res 116: 456–466.
Ardila A, Rosselli M, Strumwasser S (1991). Neuropsychological deficits in chronic cocaine abusers. Int J Neurosci 57: 73–79.
Arnold JC (2005). The role of endocannabinoid transmission in cocaine addiction. Pharmacol Biochem Behav 81: 396–406.
Arnold JM, Roberts DC (1997). A critique of fixed and progressive ratio schedules used to examine the neural substrates of drug reinforcement. Pharmacol Biochem Behav 57: 441–447.
Balster RL (1991). Drug abuse potential evaluation in animals. Br J Addict 86: 1549–1558.
Bardo MT (1998). Neuropharmacological mechanisms of drug reward: beyond dopamine in the nucleus accumbens. Crit Rev Neurobiol 12: 37–67.
Bardo MT, Rowlett JK, Harris MJ (1995). Conditioned place preference using opiate and stimulant drugs: a meta-analysis. Neurosci Biobehav Rev 19: 39–51.
Baxter MG, Chiba AA (1999). Cognitive functions of the basal forebrain. Curr Opin Neurobiol 9: 178–183.
Beatty WW, Katzung VM, Moreland VJ, Nixon SJ (1995). Neuropsychological performance of recently abstinent alcoholics and cocaine abusers. Drug Alcohol Depend 37: 247–253.
Bechara A, van der Kooy D (1989). The tegmental pedunculopontine nucleus: a brain-stem output of the limbic system critical for the conditioned place preferences produced by morphine and amphetamine. J Neurosci 9: 3400–3409.
Bechtholt AJ, Mark GP (2002). Enhancement of cocaine-seeking behavior by repeated nicotine exposure in rats. Psychopharmacology (Berl) 162: 178–185.
Berke JD, Eichenbaum HB (2001). Drug addiction and the hippocampus. Science 294: 1235.
Berlanga ML, Olsen CM, Chen V, Ikegami A, Herring BE, Duvauchelle CL et al (2003). Cholinergic interneurons of the nucleus accumbens and dorsal striatum are activated by the self-administration of cocaine. Neuroscience 120: 1149–1156.
Berlanga ML, Simpson TK, Alcantara AA (2005). Dopamine D5 receptor localization on cholinergic neurons of the rat forebrain and diencephalon: a potential neuroanatomical substrate involved in mediating dopaminergic influences on acetylcholine release. J Comp Neurol 492: 34–49.
Berridge KC, Robinson TE (1998). What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev 28: 309–369.
Bertorelli R, Consolo S (1990). D1 and D2 dopaminergic regulation of acetylcholine release from striata of freely moving rats. J Neurochem 54: 2145–2148.
Blokhina EA, Kashkin VA, Zvartau EE, Danysz W, Bespalov AY (2005). Effects of nicotinic and NMDA receptor channel blockers on intravenous cocaine and nicotine self-administration in mice. Eur Neuropsychopharmacol 15: 219–225.
Bolanos CA, Trksak GH, Cohen OS, Jackson D (2002). Differential serotonergic inhibition of in vitro striatal [3H]acetylcholine release in prenatally cocaine-exposed male and female rats. Prog Neuropsychopharmacol Biol Psychiatry 26: 1339–1348.
Bolanos CA, Trksak GH, Glatt SJ, Jackson D (2000). Prenatal cocaine exposure increases serotonergic inhibition of electrically evoked acetylcholine release from rat striatal slices at adulthood. Synapse 36: 1–11.
Bonner TI, Buckley NJ, Young AC, Brann MR (1987). Identification of a family of muscarinic acetylcholine receptor genes. Science 237: 527–532.
Bonsi P, Cuomo D, Ding J, Sciamanna G, Ulrich S, Tscherter A et al (2007). Endogenous serotonin excites striatal cholinergic interneurons via the activation of 5-HT 2C, 5-HT6, and 5-HT7 serotonin receptors: implications for extrapyramidal side effects of serotonin reuptake inhibitors. Neuropsychopharmacology 32: 1840–1854.
Bonson KR, Grant SJ, Contoreggi CS, Links JM, Metcalfe J, Weyl HL et al (2002). Neural systems and cue-induced cocaine craving. Neuropsychopharmacology 26: 376–386.
Bosboom JL, Stoffers D, Wolters E (2003). The role of acetylcholine and dopamine in dementia and psychosis in Parkinson's disease. In: R Horowski, Y Mizuno, CW Olanow, WH Poewe, P Riederer, JA Stoessl, MBH Youdim (eds). Advances in Research on Neurodegeneration, 10(Suppl 65), pp 185–195.
Bradberry CW, Barrett-Larimore RL, Jatlow P, Rubino SR (2000). Impact of self-administered cocaine and cocaine cues on extracellular dopamine in mesolimbic and sensorimotor striatum in rhesus monkeys. J Neurosci 20: 3874–3883.
Breese CR, Hampson RE, Deadwyler SA (1989). Hippocampal place cells: stereotypy and plasticity. J Neurosci 9: 1097–1111.
Browman KE, Badiani A, Robinson TE (1998). The influence of environment on the induction of sensitization to the psychomotor activating effects of intravenous cocaine in rats is dose-dependent. Psychopharmacology (Berl) 137: 90–98.
Brown EE, Robertson GS, Fibiger HC (1992). Evidence for conditional neuronal activation following exposure to a cocaine-paired environment: role of forebrain limbic structures. J Neurosci 12: 4112–4121.
Butcher SG, Butcher LLBR (1974). Origin and modulation of acetylcholine activity in the neostriatum. Brain Res 71: 167–171.
Caine SB, Koob GF, Parsons LH, Everitt BJ, Schwartz JC, Sokoloff P (1997). D3 receptor test in vitro predicts decreased cocaine self-administration in rats. NeuroReport 8: 2373–2377.
Calabresi P, Centonze D, Gubellini P, Pisani A, Bernardi G (2000). Acetylcholine-mediated modulation of striatal function. Trends Neurosci 23: 120–126.
Cambridge C (2002). Cambridge Neuropsychological Test Automated Battery Expedio (CANTABExpedio). Cambridge Cognition Limited: Cambridge, England.
Camp MC, Mayfield RD, McCracken M, McCracken L, Alcantara AA (2006). Neuroadaptations of Cdk5 in cholinergic interneurons of the nucleus accumbens and prefrontal cortex of inbred alcohol-preferring rats following voluntary alcohol drinking. Alcohol Clin Exp Res 30: 1322–1335.
Champtiaux N, Kalivas PW, Bardo MT (2006). Contribution of dihydro-beta-erythroidine sensitive nicotinic acetylcholine receptors in the ventral tegmental area to cocaine-induced behavioral sensitization in rats. Behav Brain Res 168: 120–126.
Chang JY, Sawyer SF, Lee RS, Woodward DJ (1994). Electrophysiological and pharmacological evidence for the role of the nucleus accumbens in cocaine self-administration in freely moving rats. J Neurosci 14: 1224–1244.
Chau DT, Rada P, Kosloff RA, Taylor JL, Hoebel BG (2001). Nucleus accumbens muscarinic receptors in the control of behavioral depression: antidepressant-like effects of local M1 antagonist in the Porsolt swim test. Neuroscience 104: 791–798.
Chen KC, Baxter MG, Rodefer JS (2004). Central blockade of muscarinic cholinergic receptors disrupts affective and attentional set-shifting. Eur J Neurosci 20: 1081–1088.
Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP (1999). Limbic activation during cue-induced cocaine craving. Am J Psychiatry 156: 11–18.
Ciccocioppo R, Sanna PP, Weiss F (2001). Cocaine-predictive stimulus induces drug-seeking behavior and neural activation in limbic brain regions after multiple months of abstinence: reversal by D(1) antagonists. Proc Natl Acad Sci USA 98: 1976–1981.
Clarke PB, Pert A (1985). Autoradiographic evidence for nicotine receptors on nigrostriatal and mesolimbic dopaminergic neurons. Brain Res 348: 355–358.
Colgin LL, Kubota D, Lynch G (2003). Cholinergic plasticity in the hippocampus. Proc Natl Acad Sci USA 100: 2872–2877.
Collins SL, Izenwasser S (2004). Chronic nicotine differentially alters cocaine-induced locomotor activity in adolescent vs adult male and female rats. Neuropharmacology 46: 349–362.
Consolo S, Caltavuturo C, Colli E, Recchia M, Di Chiara G (1999). Different sensitivity of in vivo acetylcholine transmission to D1 receptor stimulation in shell and core of nucleus accumbens. Neuroscience 89: 1209–1217.
Cooper DC, Klipec WD, Fowler MA, Ozkan ED (2006). A role for the subiculum in the brain motivation/reward circuitry. Behav Brain Res 174: 225–231.
Coyle JT, Price DL, DeLong MR (1983). Alzheimer's disease: a disorder of cortical cholinergic innervation. Science 219: 1184–1190.
Cragg SJ (2006). Meaningful silences: how dopamine listens to the ACh pause. Trends Neurosci 29: 125–131.
Crespo JA, Sturm K, Saria A, Zernig G (2006). Activation of muscarinic and nicotinic acetylcholine receptors in the nucleus accumbens core is necessary for the acquisition of drug reinforcement. J Neurosci 26: 6004–6010.
Crombag HS, Badiani A, Robinson TE (1996). Signalled versus unsignalled intravenous amphetamine: large differences in the acute psychomotor response and sensitization. Brain Res 722: 227–231.
Cummings JL (2000). Cholinesterase inhibitors: a new class of psychotropic compounds. Am J Psychiatry 157: 4–15.
Dackis CA, O’Brien CP (2001). Cocaine dependence: a disease of the brain's reward centers. J Substance Abuse Treat 21: 111–117.
Dalley JW, Laane K, Pena Y, Theobald DE, Everitt BJ, Robbins TW (2005). Attentional and motivational deficits in rats withdrawn from intravenous self-administration of cocaine or heroin. Psychopharmacology (Berl) 182: 579–587.
Day J, Fibiger HC (1992). Dopaminergic regulation of cortical acetylcholine release. Synapse 12: 281–286.
Day JC, Fibiger HC (1994). Dopaminergic regulation of septohippocampal cholinergic neurons. J Neurochem 63: 2086–2092.
de la Garza R, Johanson CE (1982). Effects of haloperidol and physostigmine on self-administration of local anesthetics. Pharmacol Biochem Behav 17: 1295–1299.
Deng P, Zhang Y, Xu ZC (2007). Involvement of I(h) in dopamine modulation of tonic firing in striatal cholinergic interneurons. J Neurosci 27: 3148–3156.
Desai RI, Terry P (2003). Evidence of cross-tolerance between behavioural effects of nicotine and cocaine in mice. Psychopharmacology (Berl) 166: 111–119.
Di Chiara G, Bassareo V, Fenu S, De Luca MA, Spina L, Cadoni C et al (2004). Dopamine and drug addiction: the nucleus accumbens shell connection. Neuropharmacology 47(Suppl 1): 227–241.
Di Chiara G, Tanda G, Bassareo V, Pontieri F, Acquas E, Fenu S et al (1999). Drug addiction as a disorder of associative learning. Role of nucleus accumbens shell/extended amygdala dopamine. Ann NY Acad Sci 877: 461–485.
Ding YS, Logan J, Bermel R, Garza V, Rice O, Fowler JS et al (2000). Dopamine receptor-mediated regulation of striatal cholinergic activity: positron emission tomography studies with norchloro[18F]fluoroepibatidine. J Neurochem 74: 1514–1521.
Drukarch B, Schepens E, Stoof JC (1990). Muscarinic receptor activation attenuates D2 dopamine receptor mediated inhibition of acetylcholine release in rat striatum: indications for a common signal transduction pathway. Neuroscience 37: 1–9.
Ellis JM (2005). Cholinesterase inhibitors in the treatment of dementia. J Am Osteopath Assoc 105: 145–158.
Everitt BJ, Parkinson JA, Olmstead MC, Arroyo M, Robledo P, Robbins TW (1999). Associative processes in addiction and reward. The role of amygdala-ventral striatal subsystems. Ann NY Acad Sci 877: 412–438.
Everitt BJ, Robbins TW (2005). Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci 8: 1481–1489.
Fink-Jensen A, Fedorova I, Wortwein G, Woldbye DP, Rasmussen T, Thomsen M et al (2003). Role for M5 muscarinic acetylcholine receptors in cocaine addiction. J Neurosci Res 74: 91–96.
Flynn DD, Vaishnav AA, Mash DC (1992). Interactions of cocaine with primary and secondary recognition sites on muscarinic receptors. Mol Pharmacol 41: 736–742.
Forster GL, Yeomans JS, Takeuchi J, Blaha CD (2002). M5 muscarinic receptors are required for prolonged accumbal dopamine release after electrical stimulation of the pons in mice. J Neurosci 22: RC190.
Furey ML, Pietrini P, Haxby JV (2000). Cholinergic enhancement and increased selectivity of perceptual processing during working memory. Science 290: 2315–2319.
Gardner EL (2000). What we have learned about addiction from animal models of drug self-administration. Am J Addict 9: 285–313.
Gerdeman GL, Partridge JG, Lupica CR, Lovinger DM (2003). It could be habit forming: drugs of abuse and striatal synaptic plasticity. Trends Neurosci 26: 184–192.
Goldstein RZ, Volkow ND (2002). Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry 159: 1642–1652.
Gorelick DA, Gardner EL, Xi ZX (2004). Agents in development for the management of cocaine abuse. Drugs 64: 1547–1573.
Graham AJ, Martin-Ruiz CM, Teaktong T, Ray MA, Court JA (2002). Human brain nicotinic receptors, their distribution and participation in neuropsychiatric disorders. Curr Drug Targets CNS Neurol Disord 1: 387–397.
Grecksch G, Ott T, Matthies H (1978). Influence of post-training intrahippocampally applied oxotremorine on the consolidation of a brightness discrimination. Pharmacol Biochem Behav 8: 215–218.
Grillner P, Berretta N, Bernardi G, Svensson TH, Mercuri NB (2000). Muscarinic receptors depress GABAergic synaptic transmission in rat midbrain dopamine neurons. Neuroscience 96: 299–307.
Harsing Jr LG, Zigmond MJ (1998). Postsynaptic integration of cholinergic and dopaminergic signals on medium-sized GABAergic projection neurons in the neostriatum. Brain Res Bull 45: 607–613.
Hasselmo ME, Fehlau BP (2001). Differences in time course of ACh and GABA modulation of excitatory synaptic potentials in slices of rat hippocampus. J Neurophysiol 86: 1792–1802.
Hasselmo ME, Hay J, Ilyn M, Gorchetchnikov A (2002). Neuromodulation, theta rhythm and rat spatial navigation. Neural Netw 15: 689–707.
Heidbreder CA, Shippenberg TS (1996). Evidence for an involvement of muscarinic cholinergic systems in the induction but not expression of behavioral sensitization to cocaine. Synapse 24: 182–192.
Heidbreder CA, Thompson AC, Shippenberg TS (1996). Role of extracellular dopamine in the initiation and long-term expression of behavioral sensitization to cocaine. J Pharmacol Exp Ther 278: 490–502.
Hikida T, Kaneko S, Isobe T, Kitabatake Y, Watanabe D, Pastan I et al (2001). Increased sensitivity to cocaine by cholinergic cell ablation in nucleus accumbens. Proc Natl Acad Sci USA 98: 13351–13354.
Hikida T, Kitabatake Y, Pastan I, Nakanishi S (2003). Acetylcholine enhancement in the nucleus accumbens prevents addictive behaviors of cocaine and morphine. Proc Natl Acad Sci USA 100: 6169–6173.
Hirsh R, Davis RE, Holt L (1979). Fornix—thalamus fibers, motivational states, and contextual retrieval. Exp Neurol 65: 373–390.
Horner B, Scheibe K, Stine S (1996). Cocaine abuse and attention-deficit hyperactivity disorder: implications of adult symptomatology. Psychol Addict Behav 10: 55–60.
Hyman SE (2005). Addiction: a disease of learning and memory. Am J Psychiatry 162: 1414–1422.
Hyman SE, Malenka RC, Nestler EJ (2006). Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci 29: 565–598.
Ikemoto S, Wise RA (2002). Rewarding effects of the cholinergic agents carbachol and neostigmine in the posterior ventral tegmental area. J Neurosci 22: 9895–9904.
Imperato A, Obinu MC, Gessa GL (1993a). Effects of cocaine and amphetamine on acetylcholine release in the hippocampus and caudate nucleus. Eur J Pharmacol 238: 377–381.
Imperato A, Obinu MC, Gessa GL (1993b). Stimulation of both dopamine D1 and D2 receptors facilitates in vivo acetylcholine release in the hippocampus. Brain Res 618: 341–345.
Imperato A, Obinu MC, Mascia MS, Casu MA, Zocchi A, Cabib S et al (1996). Strain-dependent effects of dopamine agonists on acetylcholine release in the hippocampus: an in vivo study in mice. Neuroscience 70: 653–660.
Ince E, Ciliax BJ, Levey AI (1997). Differential expression of D1 and D2 dopamine and m4 muscarinic acetylcholine receptor proteins in identified striatonigral neurons. Synapse 27: 357–366.
Itzhak Y, Martin JL (2000). Scopolamine inhibits cocaine-conditioned but not unconditioned stimulant effects in mice. Psychopharmacology (Berl) 152: 216–223.
Jacobs EH, Smit AB, de Vries TJ, Schoffelmeer AN (2003). Neuroadaptive effects of active versus passive drug administration in addiction research. Trends Pharmacol Sci 24: 566–573.
Jaffard R, Destrade C, Soumireu-Mourat B, Durkin T, Ebel A (1980). Changes in hippocampal cholinergic activity following learning in mice. Neurosci Lett 19: 349–352.
Janowsky DS, Overstreet DH (2000). The role of acetylcholine mechanisms in affective disorders. In: JF Mendo-Woodruff (ed). Psychopharmacology: The Fourth Generation of Progress (Retrieved July 15, 2007 from http://WWW.acnp.org/g4/GN401000095/Default.htm).
Jovanovski D, Erb S, Zakzanis KK (2005). Neurocognitive deficits in cocaine users: a quantitative review of the evidence. J Clin Exp Neuropsychol 27: 189–204.
Kalivas PW (1995). Interactions between dopamine and excitatory amino acids in behavioral sensitization to psychostimulants. Drug Alcohol Depend 37: 95–100.
Kalivas PW (2004). Glutamate systems in cocaine addiction. Curr Opin Pharmacol 4: 23–29.
Kalivas PW, Pierce RC, Cornish J, Sorg BA (1998). A role for sensitization in craving and relapse in cocaine addiction. J Psychopharmacol 12: 49–53.
Karpen JW, Hess GP (1986). Cocaine, phencyclidine, and procaine inhibition of the acetylcholine receptor: characterization of the binding site by stopped-flow measurements of receptor-controlled ion flux in membrane vesicles. Biochemistry (Mosc) 25: 1777–1785.
Katz JL, Higgins ST (2003). The validity of the reinstatement model of craving and relapse to drug use. Psychopharmacology (Berl) 168: 21–30.
Kauer JA (2003). Addictive drugs and stress trigger a common change at VTA synapses. Neuron 37: 549–550.
Kelley AE, Berridge KC (2002). The neuroscience of natural rewards: relevance to addictive drugs. J Neurosci 22: 3306–3311.
Kilts CD, Schweitzer JB, Quinn CK, Gross RE, Faber TL, Muhammad F et al (2001). Neural activity related to drug craving in cocaine addiction. Arch Gen Psychiatry 58: 334–341.
Kimura M, Yamada H, Matsumoto N (2003). Tonically active neurons in the striatum encode motivational contexts of action. Brain Dev 25(Suppl 1): S20–S23.
Kish SJ, Kalasinsky KS, Furukawa Y, Guttman M, Ang L, Li L et al (1999). Brain choline acetyltransferase activity in chronic, human users of cocaine, methamphetamine, and heroin. Mol Psychiatry 4: 26–32.
Kitabatake Y, Hikida T, Watanabe D, Pastan I, Nakanishi S (2003). Impairment of reward-related learning by cholinergic cell ablation in the striatum. Proc Natl Acad Sci USA 100: 7965–7970.
Kleber HD (2003). Pharmacologic treatments for heroin and cocaine dependence. Am J Addict 12(Suppl 2): S5–S18.
Klink R, de Kerchove d’Exaerde A, Zoli M, Changeux JP (2001). Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci 21: 1452–1463.
Koob GF (2000). Animal models of drug addiction. In: Psychopharmacology: The Fourth Generation of Progress (Retrieved July 15, 2007 from http://WWW.acnp.org/g4/GN401000072/Default.htm).
Koos T, Tepper JM (2002). Dual cholinergic control of fast-spiking interneurons in the neostriatum. J Neurosci 22: 529–535.
Kosten TR, George TP, Kosten TA (2002). The potential of dopamine agonists in drug addiction. Expert Opin Investig Drugs 11: 491–499.
Kupfermann I, Kandel ER, Iversen S (2000). Motivational and Addictive States. McGraw-Hill: New York.
Lautin A (2001). The Limbic Brain. Kluwer Academics/Plenum Publishers: New York.
Lecca D, Cacciapaglia F, Valentini V, Acquas E, Di Chiara G (2007). Differential neurochemical and behavioral adaptation to cocaine after response contingent and noncontingent exposure in the rat. Psychopharmacology (Berl) 191: 653–667.
Legault G, Smith CT, Beninger RJ (2006). Post-training intra-striatal scopolamine or flupenthixol impairs radial maze learning in rats. Behav Brain Res 170: 148–155.
Levey AI (1996). Muscarinic acetylcholine receptor expression in memory circuits: implications for treatment of Alzheimer disease. Proc Natl Acad Sci USA 93: 13541–13546.
Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR (1991). Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci 11: 3218–3226.
Lewis PR, Shute CC (1967). The cholinergic limbic system: projections to hippocampal formation, medial cortex, nuclei of the ascending cholinergic reticular system, and the subfornical organ and supra-optic crest. Brain 90: 521–540.
Lipton JW, Olsen RW, Ellison GD (1995). Length of continuous cocaine exposure determines the persistence of muscarinic and benzodiazepine receptor alterations. Brain Res 676: 378–385.
Lucki I (1997). The forced swimming test as a model for core and component behavioral effects of antidepressant drugs. Behav Pharmacol 8: 523–532.
Ma Y, Kiesewetter DO, Jagoda EM, Huang BX, Eckelman WC (2002). Identification of metabolites of fluorine-18-labeled M2 muscarinic receptor agonist, 3-(3-[(3-fluoropropyl)thio]-1,2,5-thiadiazol-4-yl)-1,2,5,6-tetrahydro-1-me thylpyridine, produced by human and rat hepatocytes. J Chromatogr B Anal Technol Biomed Life Sci 766: 319–329.
Macedo DS, Correia EE, Vasconcelos SM, Aguiar LM, Viana GS, Sousa FC (2004). Cocaine treatment causes early and long-lasting changes in muscarinic and dopaminergic receptors. Cell Mol Neurobiol 24: 129–136.
Maggos CE, Tsukada H, Kakiuchi T, Nishiyama S, Myers JE, Kreuter J et al (1998). Sustained withdrawal allows normalization of in vivo [11C]N-methylspiperone dopamine D2 receptor binding after chronic binge cocaine: a positron emission tomography study in rats. Neuropsychopharmacology 19: 146–153.
Mark GP, Blander DS, Hoebel BG (1991). A conditioned stimulus decreases extracellular dopamine in the nucleus accumbens after the development of a learned taste aversion. Brain Res 551: 308–310.
Mark GP, Hajnal A, Kinney AE, Keys AS (1999). Self-administration of cocaine increases the release of acetylcholine to a greater extent than response-independent cocaine in the nucleus accumbens of rats. Psychopharmacology (Berl) 143: 47–53.
Mark GP, Rada P, Pothos E, Hoebel BG (1992). Effects of feeding and drinking on acetylcholine release in the nucleus accumbens, striatum, and hippocampus of freely behaving rats. J Neurochem 58: 2269–2274.
Mark GP, Weinberg JB, Rada PV, Hoebel BG (1995). Extracellular acetylcholine is increased in the nucleus accumbens following the presentation of an aversively conditioned taste stimulus. Brain Res 688: 184–188.
Mayfield RD, Larson G, Zahniser NR (1992). Cocaine-induced behavioral sensitization and D1 dopamine receptor function in rat nucleus accumbens and striatum. Brain Res 573: 331–335.
McGaughy J, Dalley JW, Morrison CH, Everitt BJ, Robbins TW (2002). Selective behavioral and neurochemical effects of cholinergic lesions produced by intrabasalis infusions of 192 IgG-saporin on attentional performance in a five-choice serial reaction time task. J Neurosci 22: 1905–1913.
McIntyre CK, Ragozzino ME, Gold PE (1998). Intra-amygdala infusions of scopolamine impair performance on a conditioned place preference task but not a spatial radial maze task. Behav Brain Res 95: 219–226.
McKinney M, Coyle JT, Hedreen JC (1983). Topographic analysis of the innervation of the rat neocortex and hippocampus by the basal forebrain cholinergic system. J Comp Neurol 217: 103–121.
Meil WM, See RE (1997). Lesions of the basolateral amygdala abolish the ability of drug associated cues to reinstate responding during withdrawal from self-administered cocaine. Behav Brain Res 87: 139–148.
Mena-Segovia JRH, Magill PJ, Bolam JP (2005). The Pudunculopontine Nucleus: Towards a Functional Integration with the Basal Ganglia. Springer Science and Business Media: New York.
Mesulam MM (1996). The systems-level organization of cholinergic innervation in the human cerebral cortex and its alterations in Alzheimer's disease. Prog Brain Res 109: 285–297.
Mihailescu S, Drucker-Colin R (2000). Nicotine, brain nicotinic receptors, and neuropsychiatric disorders. Arch Med Res 31: 131–144.
Miliaressis E, Rompre PP, Laviolette P, Philippe L, Coulombe D (1986). The curve-shift paradigm in self-stimulation. Physiol Behav 37: 85–91.
Morris G, Arkadir D, Nevet A, Vaadia E, Bergman H (2004). Coincident but distinct messages of midbrain dopamine and striatal tonically active neurons. Neuron 43: 133–143.
Muir JL, Dunnett SB, Robbins TW, Everitt BJ (1992). Attentional functions of the forebrain cholinergic systems: effects of intraventricular hemicholinium, physostigmine, basal forebrain lesions and intracortical grafts on a multiple-choice serial reaction time task. Exp Brain Res 89: 611–622.
Muir JL, Everitt BJ, Robbins TW (1995). Reversal of visual attentional dysfunction following lesions of the cholinergic basal forebrain by physostigmine and nicotine but not by the 5-HT3 receptor antagonist, ondansetron. Psychopharmacology (Berl) 118: 82–92.
Muller CP, Carey RJ, Huston JP (2003). Serotonin as an important mediator of cocaine's behavioral effects. Drugs Today (Barc) 39: 497–511.
Museo E, Wise RA (1994). Place preference conditioning with ventral tegmental injections of cytisine. Life Sci 55: 1179–1186.
Niu L, Abood LG, Hess GP (1995). Cocaine: mechanism of inhibition of a muscle acetylcholine receptor studied by a laser-pulse photolysis technique. Proc Natl Acad Sci USA 92: 12008–12012.
O’Brien CP, Gardner EL (2005). Critical assessment of how to study addiction and its treatment: human and non-human animal models. Pharmacol Ther 108: 18–58.
Olds J, Milner P (1954). Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol 47: 419–427.
Olmstead MC, Franklin KB (1993). Effects of pedunculopontine tegmental nucleus lesions on morphine-induced conditioned place preference and analgesia in the formalin test. Neuroscience 57: 411–418.
Palencia CA, Ragozzino ME (2006). The effect of N-methyl-D-aspartate receptor blockade on acetylcholine efflux in the dorsomedial striatum during response reversal learning. Neuroscience 143: 671–678.
Papez J (1995). A proposed mechanism of emotion. J Neuropsychiatry Clin Neurosci 7: 103–112.
Parasuraman R (1998). The Attentive Brain. MIT Press: Cambridge, MA.
Pavlov IP (1927). Conditioned Reflexes. Routledge and Kegan Paul: London.
Pepeu G, Giovannini MG (2004). Changes in acetylcholine extracellular levels during cognitive processes. Learn Mem 11: 21–27.
Pfister M, Boix F, Huston JP, Schwarting RK (1994). Different effects of scopolamine on extracellular acetylcholine levels in neostriatum and nucleus accumbens measured in vivo: possible interaction with aversive stimulation. J Neural Transm Gen Sect 97: 13–25.
Pisani A, Bonsi P, Catania MV, Giuffrida R, Morari M, Marti M et al (2002). Metabotropic glutamate 2 receptors modulate synaptic inputs and calcium signals in striatal cholinergic interneurons. J Neurosci 22: 6176–6185.
Pisani A, Bonsi P, Picconi B, Tolu M, Giacomini P, Scarnati E (2001). Role of tonically-active neurons in the control of striatal function: cellular mechanisms and behavioral correlates. Prog Neuropsychopharmacol Biol Psychiatry 25: 211–230.
Porsolt RD, Anton G, Blavet N, Jalfre M (1978). Behavioural despair in rats: a new model sensitive to antidepressant treatments. Eur J Pharmacol 47: 379–391.
Power AE, McIntyre CK, Litmanovich A, McGaugh JL (2003). Cholinergic modulation of memory in the basolateral amygdala involves activation of both m1 and m2 receptors. Behav Pharmacol 14: 207–213.
Pratt WE, Kelley AE (2004). Nucleus accumbens acetylcholine regulates appetitive learning and motivation for food via activation of muscarinic receptors. Behav Neurosci 118: 730–739.
Pratt WE, Kelley AE (2005). Striatal muscarinic receptor antagonism reduces 24-h food intake in association with decreased preproenkephalin gene expression. Eur J Neurosci 22: 3229–3240.
Quirion R, Aubert I, Lapchak PA, Schaum RP, Teolis S, Gauthier S et al (1989). Muscarinic receptor subtypes in human neurodegenerative disorders: focus on Alzheimer's disease. Trends Pharmacol Sci (Suppl): 80–84.
Rada P, Avena NM, Hoebel BG (2005). Daily bingeing on sugar repeatedly releases dopamine in the accumbens shell. Neuroscience 134: 737–744.
Rada P, Colasante C, Skirzewski M, Hernandez L, Hoebel B (2006). Behavioral depression in the swim test causes a biphasic, long-lasting change in accumbens acetylcholine release, with partial compensation by acetylcholinesterase and muscarinic-1 receptors. Neuroscience 141: 67–76.
Rada P, Hoebel BG (2005). Acetylcholine in the accumbens is decreased by diazepam and increased by benzodiazepine withdrawal: a possible mechanism for dependency. Eur J Pharmacol 508: 131–138.
Rada P, Jensen K, Hoebel BG (2001). Effects of nicotine and mecamylamine-induced withdrawal on extracellular dopamine and acetylcholine in the rat nucleus accumbens. Psychopharmacology (Berl) 157: 105–110.
Rada P, Johnson DF, Lewis MJ, Hoebel BG (2004). In alcohol-treated rats, naloxone decreases extracellular dopamine and increases acetylcholine in the nucleus accumbens: evidence of opioid withdrawal. Pharmacol Biochem Behav 79: 599–605.
Rada P, Pothos E, Mark GP, Hoebel BG (1991). Microdialysis evidence that acetylcholine in the nucleus accumbens is involved in morphine withdrawal and its treatment with clonidine. Brain Res 561: 354–356.
Rada PV, Hoebel BG (2000). Supraadditive effect of d-fenfluramine plus phentermine on extracellular acetylcholine in the nucleus accumbens: possible mechanism for inhibition of excessive feeding and drug abuse. Pharmacol Biochem Behav 65: 369–373.
Rada PV, Hoebel BG (2001). Aversive hypothalamic stimulation releases acetylcholine in the nucleus accumbens, and stimulation-escape decreases it. Brain Res 888: 60–65.
Ragozzino ME, Choi D (2004). Dynamic changes in acetylcholine output in the medial striatum during place reversal learning. Learn Mem 11: 70–77.
Ragozzino ME, Jih J, Tzavos A (2002). Involvement of the dorsomedial striatum in behavioral flexibility: role of muscarinic cholinergic receptors. Brain Res 953: 205–214.
Rasch BH, Born J, Gais S (2006). Combined blockade of cholinergic receptors shifts the brain from stimulus encoding to memory consolidation. J Cogn Neurosci 18: 793–802.
Redgrave P, Horrell RI (1976). Potentiation of central reward by localised perfusion of acetylcholine and 5-hydroxytryptamine. Nature 262: 305–307.
Reid MS, Mickalian JD, Delucchi KL, Berger SP (1999). A nicotine antagonist, mecamylamine, reduces cue-induced cocaine craving in cocaine-dependent subjects. Neuropsychopharmacology 20: 297–307.
Reid MS, Mickalian JD, Delucchi KL, Hall SM, Berger SP (1998). An acute dose of nicotine enhances cue-induced cocaine craving. Drug Alcohol Depend 49: 95–104.
Rilling LM, Adinoff B (2007). Cognitive dysfunction in cocaine abuse: evidence for impairments in impulse control and decision-making. In: Pizzoli LA (ed). Cocaine and Heroin Abuse Research. Nova Science Publishers: Hauppauge, NY. pp 81–95.
Robbins TW (2002). The 5-choice serial reaction time task: behavioural pharmacology and functional neurochemistry. Psychopharmacology (Berl) 163: 362–380.
Roberts DC (2005). Preclinical evidence for GABAB agonists as a pharmacotherapy for cocaine addiction. Physiol Behav 86: 18–20.
Robinson SE, Hambrecht KL (1988). The effect of cocaine on hippocampal cholinergic and noradrenergic metabolism. Brain Res 457: 383–385.
Robinson TE, Berridge KC (1993). The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev 18: 247–291.
Robinson TE, Browman KE, Crombag HS, Badiani A (1998). Modulation of the induction or expression of psychostimulant sensitization by the circumstances surrounding drug administration. Neurosci Biobehav Rev 22: 347–354.
Roediger III HL (1990). Implicit memory. Retention without remembering. Am Psychol 45: 1043–1056.
Rosselli M, Ardila A, Lubomski M, Murray S, King K (2001). Personality profile and neuropsychological test performance in chronic cocaine-abusers. Int J Neurosci 110: 55–72.
Sanchis-Segura C, Spanagel R (2006). Behavioural assessment of drug reinforcement and addictive features in rodents: an overview. Addict Biol 11: 2–38.
Santiago MP, Potter LT (2001). Biotinylated m4-toxin demonstrates more M4 muscarinic receptor protein on direct than indirect striatal projection neurons. Brain Res 894: 12–20.
Sarter M, Bruno JP, Givens B (2003). Attentional functions of cortical cholinergic inputs: what does it mean for learning and memory? Neurobiol Learn Mem 80: 245–256.
Sarter M, Bruno JP, Turchi J (1999). Basal forebrain afferent projections modulating cortical acetylcholine, attention, and implications for neuropsychiatric disorders. Ann NY Acad Sci 877: 368–382.
Schacter DL (1992). Implicit knowledge: new perspectives on unconscious processes. Proc Natl Acad Sci USA 89: 11113–11117.
Schechter MD, Calcagnetti DJ (1993). Trends in place preference conditioning with a cross-indexed bibliography; 1957–1991. Neurosci Biobehav Rev 17: 21–41.
Schindler CW, Panlilio LV, Goldberg SR (2002). Second-order schedules of drug self-administration in animals. Psychopharmacology (Berl) 163: 327–344.
Schoffelmeer AN, De Vries TJ, Wardeh G, van de Ven HW, Vanderschuren LJ (2002). Psychostimulant-induced behavioral sensitization depends on nicotinic receptor activation. J Neurosci 22: 3269–3276.
Schroeder JP, Packard MG (2004). Facilitation of memory for extinction of drug-induced conditioned reward: role of amygdala and acetylcholine. Learn Mem 11: 641–647.
See RE (2005). Neural substrates of cocaine-cue associations that trigger relapse. Eur J Pharmacol 526: 140–146.
See RE, McLaughlin J, Fuchs RA (2003). Muscarinic receptor antagonism in the basolateral amygdala blocks acquisition of cocaine-stimulus association in a model of relapse to cocaine-seeking behavior in rats. Neuroscience 117: 477–483.
Seeger T, Fedorova I, Zheng F, Miyakawa T, Koustova E, Gomeza J et al (2004). M2 muscarinic acetylcholine receptor knock-out mice show deficits in behavioral flexibility, working memory, and hippocampal plasticity. J Neurosci 24: 10117–10127.
Self DW (2004). Regulation of drug-taking and -seeking behaviors by neuroadaptations in the mesolimbic dopamine system. Neuropharmacology 47(Suppl 1): 242–255.
Shaham Y, Shalev U, Lu L, De Wit H, Stewart J (2003). The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl) 168: 3–20.
Sharkey J, Ritz MC, Schenden JA, Hanson RC, Kuhar MJ (1988). Cocaine inhibits muscarinic cholinergic receptors in heart and brain. J Pharmacol Exp Ther 246: 1048–1052.
Sharp PE, McNaughton BL, Barnes CA (1985). Enhancement of hippocampal field potentials in rats exposed to a novel, complex environment. Brain Res 339: 361–365.
Shizgal P, Fulton S, Woodside B (2001). Brain reward circuitry and the regulation of energy balance. Int J Obes Relat Metab Disord 25(Suppl 5): S17–S21.
Siegal D, Erickson J, Varoqui H, Ang L, Kalasinsky KS, Peretti FJ et al (2004). Brain vesicular acetylcholine transporter in human users of drugs of abuse. Synapse 52: 223–232.
Singh HK, Ott T, Matthies H (1974). Effect of intrahippocampal injection of atropine on different phases of a learning experiment. Psychopharmacologia 38: 247–258.
Smelson DA, Roy A, Santana S, Engelhart C (1999). Neuropsychological deficits in withdrawn cocaine-dependent males. Am J Drug Alcohol Abuse 25: 377–381.
Smith JE, Co C, Yin X, Sizemore GM, Liguori A, Johnson III WE et al (2004a). Involvement of cholinergic neuronal systems in intravenous cocaine self-administration. Neurosci Biobehav Rev 27: 841–850.
Smith JE, Vaughn TC, Co C (2004b). Acetylcholine turnover rates in rat brain regions during cocaine self-administration. J Neurochem 88: 502–512.
Sousa FC, Gomes PB, Macedo DS, Marinho MM, Viana GS (1999). Early withdrawal from repeated cocaine administration upregulates muscarinic and dopaminergic D2-like receptors in rat neostriatum. Pharmacol Biochem Behav 62: 15–20.
Stewart J, Vezina P (1988). A comparison of the effects of intra-accumbens injections of amphetamine and morphine on reinstatement of heroin intravenous self-administration behavior. Brain Res 457: 287–294.
Stolerman IP, Shoaib M (1991). The neurobiology of tobacco addiction. Trends Pharmacol Sci 12: 467–473.
Stoof JC, Russchen FT, Verheijden PF, Hoogland PV (1987). A comparative study of the dopamine–acetylcholine interaction in telencephalic structures of the rat and of a reptile, the lizard Gekko gecko. Brain Res 404: 273–281.
Strickland TL, Mena I, Villanueva-Meyer J, Miller BL, Cummings J, Mehringer CM et al (1993). Cerebral perfusion and neuropsychological consequences of chronic cocaine use. J Neuropsychiatry Clin Neurosci 5: 419–427.
Sugita S, Uchimura N, Jiang ZG, North RA (1991). Distinct muscarinic receptors inhibit release of gamma-aminobutyric acid and excitatory amino acids in mammalian brain. Proc Natl Acad Sci USA 88: 2608–2611.
Suzuki T, Miura M, Nishimura K, Aosaki T (2001). Dopamine-dependent synaptic plasticity in the striatal cholinergic interneurons. J Neurosci 21: 6492–6501.
Swanson KL, Albuquerque EX (1987). Nicotinic acetylcholine receptor ion channel blockade by cocaine: the mechanism of synaptic action. J Pharmacol Exp Ther 243: 1202–1210.
Tessari M, Valerio E, Chiamulera C, Beardsley PM (1995). Nicotine reinforcement in rats with histories of cocaine self-administration. Psychopharmacology (Berl) 121: 282–283.
Thomsen M, Woldbye DP, Wortwein G, Fink-Jensen A, Wess J, Caine SB (2005). Reduced cocaine self-administration in muscarinic M5 acetylcholine receptor-deficient mice. J Neurosci 25: 8141–8149.
Tisch S, Silberstein P, Limousin-Dowsey P, Jahanshahi M (2004). The basal ganglia: anatomy, physiology, and pharmacology. Psychiatr Clin N Am 27: 757–799.
Tulving E, Schacter DL (1990). Priming and human memory systems. Science 247: 301–306.
van der Zee EA, Luiten PG (1999). Muscarinic acetylcholine receptors in the hippocampus, neocortex and amygdala: a review of immunocytochemical localization in relation to learning and memory. Prog Neurobiol 58: 409–471.
Vanderschuren LJ, Schmidt ED, De Vries TJ, Van Moorsel CA, Tilders FJ, Schoffelmeer AN (1999). A single exposure to amphetamine is sufficient to induce long-term behavioral, neuroendocrine, and neurochemical sensitization in rats. J Neurosci 19: 9579–9586.
Volkow ND, Ding YS, Fowler JS, Gatley SJ (2001). Imaging brain cholinergic activity with positron emission tomography: its role in the evaluation of cholinergic treatments in Alzheimer's dementia. Biol Psychiatry 49: 211–220.
Volkow ND, Fowler JS, Wang GJ, Hitzemann R, Logan J, Schlyer DJ et al (1993). Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse 14: 169–177.
Voorn P, Vanderschuren LJ, Groenewegen HJ, Robbins TW, Pennartz CM (2004). Putting a spin on the dorsal–ventral divide of the striatum. Trends Neurosci 27: 468–474.
Weiner DM, Levey AI, Brann MR (1990). Expression of muscarinic acetylcholine and dopamine receptor mRNAs in rat basal ganglia. Proc Natl Acad Sci USA 87: 7050–7054.
Wess J (1993). Molecular basis of muscarinic acetylcholine receptor function. Trends Pharmacol Sci 14: 308–313.
White NM (1996). Addictive drugs as reinforcers: multiple partial actions on memory systems. Addiction 91: 921–949.
Wilson JM, Carroll ME, Lac ST, DiStefano LM, Kish SJ (1994). Choline acetyltransferase activity is reduced in rat nucleus accumbens after unlimited access to self-administration of cocaine. Neurosci Lett 180: 29–32.
Wilson MC, Schuster CR (1973). Cholinergic influence on intravenous cocaine self-administration by rhesus monkeys. Pharmacol Biochem Behav 1: 643–649.
Winhusen TM, Somoza EC, Harrer JM, Mezinskis JP, Montgomery MA, Goldsmith RJ et al (2005). A placebo-controlled screening trial of tiagabine, sertraline and donepezil as cocaine dependence treatments. Addiction 100(Suppl 1): 68–77.
Wise RA (1996). Addictive drugs and brain stimulation reward. Annu Rev Neurosci 19: 319–340.
Wise RA, Rompre PP (1989). Brain dopamine and reward. Annu Rev Psychol 40: 191–225.
Wolf ME (1998). The role of excitatory amino acids in behavioral sensitization to psychomotor stimulants. Prog Neurobiol 54: 679–720.
Wyvell CL, Berridge KC (2000). Intra-accumbens amphetamine increases the conditioned incentive salience of sucrose reward: enhancement of reward ‘wanting’ without enhanced ‘liking’ or response reinforcement. J Neurosci 20: 8122–8130.
Yeomans J, Baptista M (1997). Both nicotinic and muscarinic receptors in ventral tegmental area contribute to brain-stimulation reward. Pharmacol Biochem Behav 57: 915–921.
Yeomans J, Forster G, Blaha C (2001). M5 muscarinic receptors are needed for slow activation of dopamine neurons and for rewarding brain stimulation. Life Sci 68: 2449–2456.
Yeomans JS, Kofman O, McFarlane V (1985). Cholinergic involvement in lateral hypothalamic rewarding brain stimulation. Brain Res 329: 19–26.
Zanetti L, de Kerchove D’Exaerde A, Zanardi A, Changeux JP, Picciotto MR, Zoli M (2006). Inhibition of both alpha7* and beta2* nicotinic acetylcholine receptors is necessary to prevent development of sensitization to cocaine-elicited increases in extracellular dopamine levels in the ventral striatum. Psychopharmacology (Berl) 187: 181–188.
Zeigler S, Lipton J, Toga A, Ellison G (1991). Continuous cocaine administration produces persisting changes in brain neurochemistry and behavior. Brain Res 552: 27–35.
Zhang W, Yamada M, Gomeza J, Basile AS, Wess J (2002). Multiple muscarinic acetylcholine receptor subtypes modulate striatal dopamine release, as studied with M1–M5 muscarinic receptor knock-out mice. J Neurosci 22: 6347–6352.
Zhou FM, Wilson C, Dani JA (2003). Muscarinic and nicotinic cholinergic mechanisms in the mesostriatal dopamine systems. Neuroscientist 9: 23–36.
Zhou FM, Wilson CJ, Dani JA (2002). Cholinergic interneuron characteristics and nicotinic properties in the striatum. J Neurobiol 53: 590–605.
Zocchi A, Pert A (1994). Alterations in striatal acetylcholine overflow by cocaine, morphine, and MK-801: relationship to locomotor output. Psychopharmacology (Berl) 115: 297–304.
Zubieta JK, Koeppe RA, Mulholland GK, Kuhl DE, Frey KA (1998). Quantification of muscarinic cholinergic receptors with [11C]NMPB and positron emission tomography: method development and differentiation of tracer delivery from receptor binding. J Cereb Blood Flow Metab 18: 619–631.
Acknowledgements
We thank Drs Bartley G Hoelbel, Deborah C Mash, and James E Smith for their valuable insights. We acknowledge Erica Schreffler for her assistance with the figure.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/Conflict of Interest
This work was funded, in part, by the National Institute on Drug Abuse Grant DA11434 and supported by the VA North Texas Health Care System. Disclosures (payment for professional services): Dr Adinoff has received compensation for consultation from GSK and Pain Insights; compensation for legal consultation from Parenti, Falk, Waas, Hernandez, and Cortina; Schell, Quillin, Mitchel & Cooley, LLP; Gerson Lehrman Group; Wicker, Smith, O’Hara, McCoy, Graham & Ford, PA; Conroy, Simberg, Ganon, Krevans & Abel, PA; Hershewe Law Firm, PC; and honorarium from Tarrant County Community College, American Academy of Addiction Psychiatry, Michigan State University, John Peter Smith Health Network, Medical University of South Carolina, Wayne State University, CME Inc., Temple University, and Texas Department of State Health Services. Dr Williams has received compensation for clinical work performed at the Productive Rehabilitation Institute of Dallas for Ergonomics (PRIDE) as well as his private psychology practice, Mark Williams, LLC. There are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Rights and permissions
About this article
Cite this article
Williams, M., Adinoff, B. The Role of Acetylcholine in Cocaine Addiction. Neuropsychopharmacol 33, 1779–1797 (2008). https://doi.org/10.1038/sj.npp.1301585
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301585
Keywords
This article is cited by
-
The hyperexcitability of laterodorsal tegmentum cholinergic neurons accompanies adverse behavioral and cognitive outcomes of prenatal stress
Scientific Reports (2023)
-
Chronic exposure to methadone induces activated microglia and astrocyte and cell death in the cerebellum of adult male rats
Metabolic Brain Disease (2023)
-
Cocaine-induced projection-specific and cell type-specific adaptations in the nucleus accumbens
Molecular Psychiatry (2022)
-
Novel Pharmacological Agents for the Treatment of Cocaine Use Disorder
Current Behavioral Neuroscience Reports (2022)
-
Cocaine potently blocks neuronal α3β4 nicotinic acetylcholine receptors in SH-SY5Y cells
Acta Pharmacologica Sinica (2020)
